Content uploaded by Douglas Jefferson
Author content
All content in this area was uploaded by Douglas Jefferson on Feb 11, 2016
Content may be subject to copyright.
Available via license: CC BY 4.0
Content may be subject to copyright.
Molecular and Therapeutic Characterization of
Anti-ectodysplasin A Receptor (EDAR) Agonist Monoclonal
Antibodies
*
□
S
Received for publication, June 3, 2011 Published, JBC Papers in Press, July 7, 2011, DOI 10.1074/jbc.M111.267997
Christine Kowalczyk
‡1
, Nathalie Dunkel
‡1,2
, Laure Willen
‡
, Margret L. Casal
§
, Elizabeth A. Mauldin
§
, Olivier Gaide
¶
,
Aubry Tardivel
‡
, Giovanna Badic
‡3
, Anne-Lise Etter
储4
, Manuel Favre
储5
, Douglas M. Jefferson**
‡‡
, Denis J. Headon
§§
,
Ste´phane Demotz
储¶¶6
, and Pascal Schneider
‡7
From the
‡
Department of Biochemistry, University of Lausanne, CH-1066 Epalinges, Switzerland, the
§
School of Veterinary
Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6010, the
¶
Department of Dermatology, University of
Geneva, CH-1211 Geneva, Switzerland,
储
Apoxis SA, CH-1004 Lausanne, Switzerland, **Cell Essential, Boston, Massachusetts 02116,
the
‡‡
Tufts University School of Medicine, Boston, Massachusetts 02111, the
§§
Roslin Institute and Royal (Dick) School of Veterinary
Studies, University of Edinburgh, Roslin EH25 9PS, Scotland, United Kingdom, and the
¶¶
Edimer Biotech, Ch de l’Eglise 7,
CH-1066 Epalinges, Switzerland
The TNF family ligand ectodysplasin A (EDA) and its receptor
EDAR are required for proper development of skin appendages
such as hair, teeth, and eccrine sweat glands. Loss of function
mutations in the Eda gene cause X-linked hypohidrotic ectoder-
mal dysplasia (XLHED), a condition that can be ameliorated in
mice and dogs by timely administration of recombinant EDA. In
this study, several agonist anti-EDAR monoclonal antibodies
were generated that cross-react with the extracellular domains
of human, dog, rat, mouse, and chicken EDAR. Their half-life in
adult mice was about 11 days. They induced tail hair and sweat
gland formation when administered to newborn EDA-deficient
Tabby mice, with an EC
50
of 0.1 to 0.7 mg/kg. Divalency was nec-
essary and sufficient for this therapeutic activity. Only some anti-
bodies were also agonists in an in vitro surrogate activity assay
based on the activation of the apoptotic Fas pathway. Activity in
this assay correlated with small dissociation constants. When
administered in utero in mice or at birth in dogs, agonist antibodies
reverted several ectodermal dysplasia features, including tooth
morphology. These antibodies are therefore predicted to effi-
ciently trigger EDAR signaling in many vertebrate species and will
be particularly suited for long term treatments.
The TNF family includes 19 ligands, most of which control
development, function and/or homeostasis of the immune sys-
tem (1). In this respect, ectodysplasin A (EDA)
8
is an exception
as it participates in ectodermal appendage formation (2). The
Eda gene on the X chromosome is transcribed as multiple splice
variants, only two of which code for the receptor-binding C-ter-
minal TNF homology domain. These two variants, generated
by splicing at an alternative donor site between exons 8 and 9,
code for 391- and 389-amino acid-long proteins called EDA1
and EDA2 (3). EDA1 binds EDAR, whereas EDA2 binds to
another receptor, XEDAR (3). The biology of EDA2 and
XEDAR is distinct from that of EDA1. Indeed, XEDAR-defi-
cient mice have no obvious ectodermal dysplasia phenotype,
whereas mice deficient in EDA, EDAR, or the signaling adaptor
protein EDARADD all display virtually indistinguishable ecto-
dermal dysplasia phenotypes, indicating the predominance of
the EDA1-EDAR axis in the development of skin-derived
appendages (4– 8).
In humans, EDA1 loss of function mutations cause X-linked
hypohidrotic ectodermal dysplasia (XLHED), a rare condition
characterized by defective formation of teeth, hair, sweat glands
and other glands (6). Because of their insufficient number of
sweat glands, these patients are prone to hyperthermia. They
also frequently suffer from recurrent respiratory tract infec-
tions caused by abnormal mucus production in the airways.
Other problems are oligodontia, dry skin, and dry eyes (9–11).
EDA1 is a transmembrane type II protein with a furin con-
sensus cleavage site, a collagen-like domain, and a C-terminal
TNF homology domain, any of which when mutated can cause
XLHED (12). To be active, EDA must be processed and bind
EDAR through its trimeric C-terminal domain. The signaling
ability of EDA1 is re-enforced by its collagen domain that cross-
links individual EDA1 trimers (13). Interestingly, some EDA1
mutations can also cause selective tooth agenesis, a condition
characterized by no or very little involvement of other ectoder-
mal appendages (14). In these patients, EDA1 mutants retain
partial binding to EDAR, suggesting that tooth development is
particularly sensitive to “high quality” EDAR signals.
*This work was supported by grants from the Swiss National Science Foun-
dation (to P. S.) and by National Institutes of Health Grant RR02512 (to
M. L. C.). P. S., M. L. C., O. G., and S. D. are shareholders of EdimerPharma.
□
S
The on-line version of this article (available at http://www.jbc.org) contains
supplemental Figs. S1–S8.
The nucleotide sequence(s) reported in this paper has been submitted to the Gen-
BankTM/EBI Data Bank with accession number(s) JN099705 to JN099733.
1
Both authors contributed equally to this work.
2
Present address: Dept. of Oncology, Geneva University Hospital, CH-1211
Geneva, Switzerland.
3
Present address: Instrumat AG, Rueyre 116, CH-1020 Renens, Switzerland.
4
Present address: Celgene International, CH-2074 Marin, Switzerland.
5
Present address: Merck-Serono, CH-1809 Fenil-sur-Corsier, Switzerland.
6
Present address: Philip Morris Products SA, CH-2000 Neuchaˆtel, Switzerland.
7
To whom correspondence should be addressed. Tel.: 41-21-692-5709; Fax:
41-21-692-5705; E-mail: pascal.schneider@unil.ch.
8
The abbreviations used are: EDA, ectodysplasin A; EDAR, EDA receptor;
XLHED, X-linked hypohidrotic ectodermal dysplasia.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 286, NO. 35, pp. 30769–30779, September 2, 2011
© 2011 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30769
by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from by guest on January 13, 2016http://www.jbc.org/Downloaded from
Transgenic expression of EDA1 in skin under the keratin 14
promoter results in a disheveled hair phenotype, hypertrophy
of sebaceous glands, and formation of supernumerary molars
or nipples (15). Transgenic EDA1 expression in the skin of
EDA-deficient Tabby mice corrected many of the ectodermal
dysplasia defects (16). The reverted phenotype was stable even
after shutdown of transgenic EDA1 expression in young adults,
suggesting that EDA1 plays a role in the formation but not in
the maintenance of skin appendages. Interruption of EDA1
expression, however, resulted in the normalization of seba-
ceous gland size (16). Similar conclusions were reached with an
alternative approach of protein replacement therapy, in which
EDA-deficient animals were exposed to a recombinant form of
EDA during development (17, 18). Taken together, these data
provide a proof of concept for protein replacement therapy in
young patients with XLHED.
In this study, we generated agonist anti-EDAR antibodies
that mimic the action of transgenic or recombinant EDA1 in
development. Most of these antibodies cross-react with EDAR
of mammals and birds and are active as monomeric, divalent
molecules. They corrected, among others, sweat glands, tra-
cheal glands, and tooth morphology in EDA-deficient mice and
were also active in EDA-deficient dogs. These mouse monoclo-
nal antibodies will be reagents of choice for long term experi-
ments in mice and pave the way for the development of thera-
peutic antibodies for use in XLHED or other EDAR-related
applications in humans.
EXPERIMENTAL PROCEDURES
Animals—Mice were handled according to Swiss Federal
Veterinary Office guidelines, under the authorization of the
Office Ve´te´rinaire Cantonal du Danton de Vaud (authorization
1370.3 to P. S.). White-bellied agouti B6CBAa A
w⫺J
/A-Eda
Ta
/J
Tabby mice (000314; The Jackson Laboratory) were bred as
Eda
Ta
/Eda
Ta
and Eda
Ta
/Y mutants or as ⫹/⫹and ⫹/Y wild
type controls. EDAR-deficient OVE1B mice were as described
previously (5). EDA-deficient dogs (19) were cared for in
accordance with the principles outlined in the National Insti-
tutes of Health Guide for the Care and Use of Laboratory Ani-
mals and in the International Guiding Principles for Biomedical
Research Involving Animals.
Plasmids and Recombinant Proteins—Plasmids used in this
study were either previously published or derived from the pub-
lished plasmids by standard molecular biology techniques (sup-
plemental Fig. S1) (13, 20, 21). A fully human form of Fc-EDA1
was kindly provided by Dr. Neil Kirby (EdimerPharma, Boston).
hEDAR-Fc and mEDAR-Fc were produced and purified as
described previously (21).
Generation and Purification of Anti-EDAR Monoclonal
Antibodies—150
g of hEDAR-Fc or mEDAR-Fc (amino acid
residues 29–183 supplemental Fig. S1) were briefly sonicated
three times in 750
l of PBS/STIMUNE (1:1, v/v) (Cedi-diag-
nostics, Lelystad, The Netherlands). Female OVE1B mice were
immunized subcutaneously (base of tail, 200
l) with the anti-
gen preparation and boosted between days 10 and 14 with anti-
gen in PBS/STIMUNE (base of tail, 150
l). Mice positive for
anti-EDAR antibodies at day 30 were boosted with 150
gof
antigen in PBS at day 40 (base of tail). Three days later, lymph
node cells were fused with myeloma cells according to standard
procedures, grown in complete RPMI 1640 medium over a
feeder layer of mouse macrophages, and selected 24 h later
with hypoxanthine/aminopterin/thymidine-containing medium.
Supernatants of 96-well plates were tested by ELISA for antibody
secretion. Positive clones were subcloned twice by limiting dilu-
tion and then slowly adapted to medium without macrophages
and hypoxanthine/aminopterin/thymidine medium supplement.
Most hybridomas could then be progressively adapted to serum-
free Opti-MEM medium (Invitrogen). Antibodies were purified
from conditioned Opti-MEM supernatants by affinity chromatog-
raphy on protein G-Sepharose (GE Healthcare).
Transfections—HEK 293T cells were grown in DMEM, 10%
fetal calf serum and transfected by the calcium phosphate
method. Cells were grown for 7 days in serum-free Opti-MEM
medium (Invitrogen) for the production of EDAR-Fc trunca-
tion mutant fusion proteins or for 48 h in complete medium for
surface expression of receptors-TRAILR3 fusion proteins.
ELISA—For the detection of anti-EDAR antibodies, ELISA
plates were coated with hEDAR-Fc at 1
g/ml, blocked, and
revealed with anti-EDAR antibodies (adequately diluted serum
of EDAR immunized mice, hybridoma supernatants, or puri-
fied antibody) followed by a peroxidase-coupled goat anti-
mouse IgG (Jackson ImmunoResearch). For isotype determina-
tion, ELISA plates were coated with 1
g/ml of anti-EDAR
antibodies and revealed with peroxidase-coupled antibodies
against the heavy chain of mouse IgG1, IgG2a, or IgG2b (South-
ern Biotech). For epitope mapping, ELISA plates were coated
with an F(ab⬘)
2
fragment of a goat anti-human Ig (Jackson
ImmunoResearch) to capture various EDAR-Fc constructs or a
control receptor (B cell maturation antigen-Fc) in cell superna-
tants. EDAR-Fc constructs and B cell maturation antigen-Fc
were revealed either with peroxidase-coupled donkey anti-hu-
man (H⫹L) (Jackson ImmunoResearch) or with anti-EDAR
antibodies at 1
g/ml followed by peroxidase-coupled goat
anti-mouse IgG (Jackson ImmunoResearch) or with FLAG-
EDA1 or FLAG-BAFF followed by biotinylated anti-FLAG M2
antibody (Sigma) and peroxidase-coupled streptavidin.
SDS-PAGE, Western Blot, and Native Gel Electrophoresis—
Anti-EDAR antibodies (10
g/lane) were analyzed by SDS-
PAGE under reducing conditions followed by Coomassie Blue
staining. Antibodies were also analyzed by native gel electro-
phoresis (4
g/lane) (Biomidi, Toulouse, France) and stained
with Amido Black according to manufacturer’s instructions,
except that the electrophoresis was performed for 1 h.
To test the ability of anti-EDAR antibodies to recognize
denatured EDAR, 2
g of bovine serum albumin, 20 ng of
hEDAR-Fc, and 20 ng of hFas-Fc were analyzed by SDS-
PAGE and Western blot under reducing (100 mMdithiothre-
itol) or nonreducing conditions, revealed with the various
anti-EDAR antibodies at 1
g/ml followed by peroxidase-
coupled anti-mouse antibody (1:10,000) and ECL reagent
(GE Healthcare).
Sequencing of Anti-EDAR Antibodies—RNA was extracted
from hybridoma cells with an RNeasy kit (Qiagen) according
to the manufacturer’s guidelines. cDNA was prepared by
reverse transcription with Ready-To-Go T-Primed first-
Strand kit (GE Healthcare). Variable sequences of the heavy
Agonist Anti-EDAR Antibodies
30770 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 •NUMBER 35 •SEPTEMBER 2, 2011
by guest on January 13, 2016http://www.jbc.org/Downloaded from
and light chains were amplified by PCR as described previ-
ously (22). PCR products were sequenced on both strands.
Sequences were analyzed for gene usage using the IMGT
sequence alignment software.
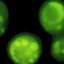
FACS Analyses—293T cells co-transfected with enhanced
GFP, and receptor-glycosylphosphatidylinositol expression
plasmids were stained with Fc-EDA1 or Fc-EDA2 essentially as
described before (20) or with anti-EDAR antibodies at 4
g/ml
followed by phycoerythrin-coupled anti-mouse secondary anti-
body. Following staining, cells were analyzed using a FACScan
flow cytometer (BD Biosciences) and FlowJo software (TreeStar,
Ashland, OR).
Fab Generation and Affinity Determination by Surface Plas-
mon Resonance—An IgG1 Fab and F(ab⬘)
2
preparation kit was
used according to the manufacturer’s instructions (Pierce).
Briefly, purified anti-EDAR antibodies were digested for 72 h at
37 °C with immobilized ficin. Fc fragments and undigested
antibodies were removed by chromatography on protein A.
The flow-through, containing the Fab and F(ab⬘)
2
fragments,
was concentrated and applied onto a Superdex-200 gel perme-
ation chromatography column eluted in PBS. Absorbance was
recorded at 280 nm.
For surface plasmon resonance, human EDAR-Fc was cap-
tured on anti-human IgG Fc-derivatized CM5 chips in a Bia-
core T100 apparatus (GE Healthcare). Fab solutions of anti-
EDAR antibodies at the indicated concentration in PBS were
applied for 90 s at 50
l/min and subsequently washed with
buffer. All curve fittings were performed assuming a 1:1 binding
model, although two antibodies had a biphasic dissociation that
did not fit the 1:1 model.
In Vitro Cytotoxicity Assays—Fas-deficient hEDAR:Fas Jur-
kat cells have been described before, and mEDAR:Fas-express-
ing cells were obtained by retroviral infection according to the
same protocol (13). The cytotoxicity assay using EDAR:Fas Jur-
kat cells was performed as described for FasL on Jurkat cells
(23).
Injections in EDA-deficient Animals—Tabby pups were
labeled by puncture of a footpad with a 30-gauge needle dipped
in china ink. Intraperitoneal injections with anti-EDAR anti-
bodies or Fc-EDA1 were performed within 24 h after birth with
a maximal volume of 15
l, using 0.5-ml U-100 insulin syringes
(BD Biosciences). Examination and photography of tail hairs
were performed at days 20–22 post-injection. Pregnant Tabby
mice were treated intravenously at days 13 and 20 (E13/E20) or
9 and 17 (E9/E17) of gestation with 400
g of anti-EDAR anti-
body (antibody 3). Offspring were analyzed at 6 months of age,
essentially as described previously (18). Age-matched wild type
and EDA-deficient Tabby mice were similarly analyzed for
comparison. Tracheal glands were detected by Alcian blue
staining (24). Three dogs affected with X-linked ectodermal
FIGURE 1. Purity of protein-G purified anti-EDAR monoclonal antibodies.
Anti-EDAR monoclonal antibodies were purified by protein G affinity chroma-
tography from culture supernatants in serum-free medium (antibodies 1– 4,
6 –10, and 12–14) or in serum-containing medium (antibodies 5, 11, and 15).
A, SDS-PAGE analysis and Coomassie Blue staining of 10
g of antibody per
lane under reducing conditions. Migration positions of molecular mass stan-
dards (in kDa) are shown. B, native gel electrophoresis of 4
g of anti-EDAR
antibodies per lane, stained with Amido Black.
TABLE 1
Characteristics of anti-EDAR monoclonal antibodies
IGHV and IGLV indicate immunoglobulin heavy and light chain variable region genes likely used in the antibody. hu is human EDAR-Fc; mu is mouse EDAR-Fc.I⫹II
indicates EDAR-Fc containing CRD1 and CRD2 of human EDAR. I is EDAR-Fc containing CRD1 only. Full is EDAR-Fc containing the full extracellular domain of EDAR.
k
a
is the association constant. k
d
is the dissociation constant. K
D
is the affinity of monomeric Fab to EDAR-Fc. EC
50
tail hair indicates the dose of antibody required to get
half-maximal tail hair reversion score when administered intraperitoneally in newborn Tabby mice. EC
50
, mEDAR:Fas or hEDAR:Fas means dose of antibody required to
kill half of the EDAR:Fas-expressing Jurkat cells.
Anti-EDAR IGHV gene IGLV gene Antigen Isotype Epitope k
a
䡠10
ⴚ5
k
d
䡠10
4
K
D
EC
50
Tail hair mEDAR:Fas hEDAR:Fas
M
⫺1
s
⫺1
s
⫺1
nMmg/kg ng/ml ng/ml
1 5–17 10–96 hu IgG1 I ⫹II 6.79 3.69 0.54 0.125 10 200
2 1–69 1–117 hu IgG1 I ⫹II 13.2 13.3 1.00 0.42 ⬎4000 ⬎4000
3 5–17 10–96 hu IgG1 I ⫹II ND ND ND 0.18 30 100
4 ND 1–117 hu IgG1 I ⫹II 4.50 13.6 3.01 0.7 ⬎4000 ⬎4000
5 2–2 4–91 hu IgG2b I ND ND ND 0.25 ⬎4000 ⬎4000
6 1S135 1–110 hu IgG2a I 3.81 6.02 1.58 0.25 ⬎4000 ⬎4000
7 7–3 4–577 hu IgG1 I ⫹II 2.92 96.6 33.1 0.3 ⬎4000 ⬎4000
8 5–17 10–96 mu IgG1 I ⫹II 8.12 3.86 0.48 0.5 100 ⬎4000
9 1–63 1–117 mu IgG1 I⫹II⫹III 1.89 8.44 4.46 0.42 ⬎4000 ⬎4000
10 1–39 4–55 hu IgG1 I ⫹II 0.35 2.38 6.78 0.125 10 50
11 1–39 10–94 hu IgG1 I ⫹II ND ND ND 3.3 ⬎4000 ⬎4000
12 1–14 17–12 mu IgG1 I 0.41 6.66 16.3 0.35 10 5
13 1S135 1–110 hu IgG1 I 1.83 18.7 10.2 0.42 ⬎4000 ⬎4000
14 1–42 10–94 mu IgG1 I ⫹II 1.90 74.8 39.3 0.3 ⬎4000 ⬎4000
15 1–63 12–44 hu IgG1 I ⫹II ND ND ND ND ND ND
Agonist Anti-EDAR Antibodies
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30771
by guest on January 13, 2016http://www.jbc.org/Downloaded from
dysplasia were administered agonist anti-EDAR antibody 3 in
the jugular vein at 2 days of life (n⫽2; 10 mg/kg) or at 14 days
of life (n⫽1; 7 mg/kg). The analysis was performed essentially
as described previously (17, 25). In particular, the dogs were
monitored daily for adverse reactions, overall health, and spe-
cific ocular and respiratory diseases. Complete blood cell
counts and serum biochemistry screens were evaluated within
2–7 days after injection with agonist anti-EDAR antibody. Den-
tal radiographs were obtained when the dogs were adults, when
about 1 year old. Shirmer tear testing was performed at
6-month intervals. Complete necropsies were performed
between 1.6 and 2.4 years of age. Tissues were fixed in 10%
neutral buffered formalin, routinely processed, sectioned at 5
m, and stained with hematoxylin and eosin.
RESULTS
Generation and Screening of Agonist Anti-EDAR Antibodies—
To obtain cross-reacting antibodies against conserved EDAR
regions, EDAR-deficient mice were immunized with either
human or mouse EDAR-Fc fusion proteins. The EDAR-defi-
cient mouse strains Downless and Sleek have loss-of-function
mutations in the extracellular or intracellular domains of
EDAR, respectively, but still express the protein. We therefore
immunized OVE1B mice in which the Edar gene is completely
FIGURE 2. Epitope mapping of anti-EDAR monoclonal antibodies. A, schematic linear representation of human EDAR showing the position of cysteine
residues (thin horizontal lines), the putative N-linked glycosylation site (thick horizontal line, N), and the six structural modules (rectangles with rounded corners)
composing the three cysteine-rich domains (CRD1, CRD2, and CRD3). The transmembrane domain (TMD), signal peptide (Leader), stalk, and intracellular
domain (ID) are also shown. Amino acid numbers at the junctions of interest are indicated. The arrowhead indicates the predicted cleavage site of the signal
peptide. The scheme is drawn to scale, except for the intracellular domain. Ctrl, control. B, the indicated EDAR-Fc constructs or B cell maturation antigen-Fc
(BCMA-Fc) control were captured in an ELISA plate and revealed with the indicated anti-EDAR antibodies, with an anti-human IgG to control efficient capture
of the various EDAR-Fc proteins, or with FLAG-EDA1 or FLAG-BAFF as controls. C, bovine serum albumin (B; BSA, 2
g), hEDAR-Fc (E, 20 ng), and hFas-Fc (F,20
ng) were resolved by SDS-PAGE under reducing or nonreducing conditions, transferred onto nitrocellulose, and probed with anti-EDAR1–15.
Agonist Anti-EDAR Antibodies
30772 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 •NUMBER 35 •SEPTEMBER 2, 2011
by guest on January 13, 2016http://www.jbc.org/Downloaded from
deleted by random genomic integration of an unrelated trans-
gene (5, 26). Hybridoma supernatants with anti-EDAR reactiv-
ity were screened for agonist activity using two complementary
tests. In the first one, surrogate reporter cell lines stably
expressing hEDAR:Fas or mEDAR:Fas fusion proteins were
used. EDAR activation in these reporter cells leads to apoptotic
cell death by activation of the Fas pathway (13). In the second
assay, hybridoma supernatants were administered intraperito-
neally to newborn, EDA-deficient Tabby pups (18). Tabby mice
completely lack tail hairs, and the agonist activity of antibodies
can therefore be measured by the induction of tail hair, pro-
vided that antibodies recognize and activate mouse EDAR. Sev-
eral hybridoma secreted agonist antibodies with in vivo activity,
but only a few were also active in the cell-based in vitro assay
(data not shown). Selected hybridoma were subcloned and
adapted for growth in serum-free medium from which antibod-
ies were purified (Fig. 1A). The monoclonal nature of these
antibodies was further confirmed by their sharp migration in
native protein electrophoresis (Fig. 1B).
Agonist Anti-EDAR Antibodies Have Varied but Relatively
Limited Sequence Characteristics—Variable regions of heavy
and light antibody chains were amplified by RT-PCR and
sequenced for several hybridoma. A number of different vari-
able region genes were identified for both the heavy and light
chains (Table 1), but some of them were shared by two or three
hybridoma, usually with different somatic mutations. Interest-
ingly, antibodies with identical heavy and light variable genes
sharing greater than 90% sequence identity were obtained from
different mice immunized with mouse (antibody 8) or human
EDAR (antibodies 1 and 3) (Table 1 and supplemental Fig. S2).
Thus, different variable genes can be used to generate agonist
anti-EDAR antibodies, but the gene repertoire must be limited
as similar antibodies were found two or three times in the rela-
tively limited panel that we have analyzed.
Agonist Antibodies Recognize at Least Three Different
Epitopes in EDAR—EDAR contains three cysteine-rich do-
mains in its extracellular region, plus a stalk sequence (Fig. 2A).
We used EDAR constructs containing these four regions alone
or in combinations to roughly characterize the epitopes recog-
nized by the antibodies. Some antibodies recognized CRD1
alone, others recognized CRD1 and -2 together, and one
reacted with CRD1–3, but none bound to receptors lacking
CRD1 (Fig. 2Band Table 1). All antibodies recognized recom-
binant EDAR by Western blot under nonreducing conditions,
but only three reacted with reduced EDAR (Fig. 2C). These
three antibodies had their epitopes localized in CRD1. It is pos-
sible that the presence of CRD1 is required for correct folding of
EDAR. Abnormal disulfide bridge formation in the absence of
FIGURE 3. Cross-species specificity of anti-EDAR antibodies. Receptors fused to the glycosylphosphatidylinositol anchor of TRAILR3 were expressed in 293T
cells together with an enhanced GFP tracer (xaxis) and stained with Fc-EDA1, Fc-EDA2, or with anti-EDAR antibodies (yaxis). Receptor expression was
confirmed by staining with an antibody directed against the C-terminal portion of TRAIL-R3 present in all constructs (mAb572). Both scattergram axes show
fluorescence intensity on a logarithmic scale (10
0
–10
4
).
Agonist Anti-EDAR Antibodies
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30773
by guest on January 13, 2016http://www.jbc.org/Downloaded from
CRD1 would explain why antibodies did not recognize con-
structs containing CRD2 and CRD3. Taken together, these
results indicate that agonist anti-EDAR antibodies can recog-
nize at least three different EDAR epitopes located in CRD1 and
probably CRD2 and CRD3.
Agonist Anti-EDAR Antibodies Cross-react with EDAR of
Various Mammals and Birds—Most anti-EDAR antibodies
cross-reacted with human, dog, rat, mouse, and chicken
EDAR when these were expressed as glycosylphosphatidyli-
nositol-anchored molecules in 293T cells (Fig. 3). Antibody 5
only reacted minimally with chicken EDAR, whereas anti-
body 15 that had been selected on the basis of its specificity
for human EDAR rather than for its agonist activity recog-
nized human and dog EDAR, but not rat, mouse and chicken
EDAR (Fig. 3).
Binding Characteristics of Anti-EDAR Antibodies to EDAR—
The affinity of 11 agonist antibodies to human EDAR was
determined by surface plasmon resonance. For this purpose,
monomeric Fab fragments were generated by ficin digestion
and size exclusion chromatography (Fig. 4A). Affinities var-
ied from 0.5 to 40 nM, and differences were also observed in
the association and dissociation constants (Fig. 4Band Table
1). The dissociation kinetics of two antibodies (7 and 14)
were biphasic, showing first a rapid dissociation followed by
a slower dissociation, but as these antibodies had no remark-
able agonist activity, this was not analyzed further.
Comparison of the Activity of Agonist Anti-EDAR Antibodies
in Vitro and in Vivo—As we have not yet been able to identify a
simple and quantitative assay to characterize EDAR agonists
using EDAR’s own signaling pathway in vitro, we used a surro-
gate reporter assay in which Fas-sensitive cells were transfected
with the extracellular domain of human or mouse EDAR fused
to the intracellular portion of Fas. Binding of an active recom-
binant EDA1 (Fc-EDA1) to these cells induces cell death by
activation of the pro-apoptotic Fas pathway (13). Interestingly,
only some of the antibodies (1, 3, 8, 10, and 12) (Fig. 5A) killed
mEDAR:Fas-expressing cells, and even fewer killed hEDAR:Fas
expressing cells (1, 3, 10, and 12) (Fig. 5Band Table 1). In all
cases, antibodies were less active than Fc-EDA1 by 1–2 orders
of magnitude. The picture was different in an in vivo assay,
where newborn Tabby mice were treated with antibodies on the
day of birth. In this case, all antibodies rescued tail hair forma-
tion in a dose-dependent manner and with similar EC
50
values
of 0.1 to 0.7 mg/kg (Fig. 6). Only one of the antibodies (antibody
11) seemed less active, with an EC
50
value of about 3 mg/kg.
Functional sweat glands were induced by the treatment with
similar EC
50
as for tail hair (data not shown). The half-life of
two antibodies (1 and 3) was determined in adult Tabby mice
and found to be 10.5 and 11 days, respectively (supplemental
Fig. S3).
Divalent Monomeric Agonist Antibodies Are Active in Vivo—
We have shown previously that cross-linked EDA1 containing
more than one trimer in a single molecule are better agonists
than trimeric EDA1 (13). We therefore wondered whether the
in vivo activity of agonist anti-EDAR antibodies was due to
monomeric antibodies or to aggregates thereof. The activity of
a monomeric (divalent) antibody purified by size exclusion
chromatography (Fig. 6, antibody 1) was, however, very similar
to that of the total preparation (data not shown), and compared
favorably (EC
50
⬃0.1 mg/kg) with recombinant Fc-EDA1 (EC
50
⬃0.05 mg/kg) (Fig. 6). In addition, the F(ab⬘)
2
fragment, but not
the Fab fragment, was active in vivo when administered to new-
born pups on the day of birth (supplemental Fig. S4). When the
F(ab⬘)
2
was administered later at day 3 post-birth, one of the
latest time points where ventral tail hair can be induced, its
action could be inhibited by an excess of the Fab fragment,
ruling out that the lack of agonist activity of the Fab would be
due only to a shorter half-life in vivo (supplemental Fig. S4). We
FIGURE 4. Generation and binding characteristics of anti-EDAR Fab fragments. A, Superdex-200 gel permeation chromatography elution profiles of intact
and ficin-digested anti-EDAR antibodies. In the digested antibodies, Fc fragments and undigested antibodies were first removed by chromatography on
protein A. Peak identities are indicated. B, Fab fragments at the indicated concentrations were analyzed by surface plasmon resonance onto immobilized
hEDAR-Fc. Fab solutions were applied for 90 s, and subsequently washed with buffer. Results for three antibodies with distinct binding characteristics are
shown.
Agonist Anti-EDAR Antibodies
30774 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 •NUMBER 35 •SEPTEMBER 2, 2011
by guest on January 13, 2016http://www.jbc.org/Downloaded from
conclude from these observations that a divalent agonist anti-
EDAR antibody is both sufficient and necessary to exert activity
in Tabby mice.
Effective Treatment of EDA-deficient Mice with Agonist Anti-
EDAR Antibodies—Some patients with partially inactivating
EDA mutations have teeth defects but otherwise normal skin
appendages, suggesting that tooth formation may require more
stringent EDAR signals than other skin appendages for proper
development (14). To test the effect of agonist antibodies on
tooth development, pregnant Tabby mothers were treated dur-
ing pregnancy so that the antibody could be transferred to
embryos by the trans-placental antibody transport system.
Mice exposed to agonist anti-EDAR during development not
only had tail hairs and functional sweat glands, but also hair
behind the ears, mucus-secreting glands in the trachea, and a
normalized eye appearance (Fig. 7, A–G, and supplemental Fig.
S5). In addition, molars of treated mice were reverted and
almost indistinguishable from those of wild type animals (Fig. 7,
Hand I, and supplemental Fig. S5). The effect was long lasting,
as a similarly treated mouse was still reverted after more than 2
years (supplemental Fig. S6). Taken together, these results indi-
cate that the two agonists anti-EDAR antibodies tested in this
application (antibodies 1 and 3, Fig. 7, supplemental Fig. S4, and
data not shown) revert the ectodermal dysplasia phenotypes
that we have looked at in Tabby mice, including tooth
morphology.
Activity of Agonist Anti-EDAR Antibodies in EDA-deficient
Dogs—Agonist anti-EDAR antibodies recognize EDAR of dif-
ferent species (Fig. 3). To test whether the observed cross-spe-
cies reactivity also holds true for the agonist activity, anti-EDAR
antibody 3 was administered intravenously to three EDA-defi-
cient dogs at either 2 days of life (n⫽2, 10 mg/kg) or at 14 days
of life (n⫽1, 7 mg/kg). None of these dogs showed adverse
reactions upon injection. Dentition was corrected not only in
FIGURE 5. In vitro activity of anti-EDAR antibodies in a surrogate reporter assay. A, anti-EDAR antibodies and Fc-EDA1 were tested for their capacity to
induce apoptosis of mEDAR:Fas-expressing Jurkat cells. After overnight culture, cell viability was determined by a cell viability assay. EC
50
are indicated as
dotted lines. Anti-EDAR antibody number (or Fc-EDA1) for each curve is indicated to the right of each panel. B, as above, except that hEDAR:Fas expressing cells
were used.
FIGURE 6. Therapeutic doses of the anti-EDAR antibodies in newborn Tabby mice. Newborn Tabby mice were injected intraperitoneally during the first 24 h
of life with graded doses of anti-EDAR antibodies, an irrelevant mouse IgG1 (Aprily 5) or Fc-EDA1. Four to 6 weeks later (antibody 2– 4) or 3 weeks later
(antibodies 1, 4 –14, and Aprily 5), hair density on the tail was scored according to the criteria shown in the inset. The anti-EDAR antibody 1 used in this
experiment was the monomer peak obtained by gel filtration (see Fig. 4).
Agonist Anti-EDAR Antibodies
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30775
by guest on January 13, 2016http://www.jbc.org/Downloaded from
EDA-deficient dogs that were treated at 2 days of life but also in
the affected treated at 14 days of life, although the latter still
lacked premolars, accounting for the decreased number of
teeth (Fig. 8 and Table 2). Interestingly, the premolars and
molars in dogs treated at day 2 of life had a more normal
appearance than in those dogs treated with Fc-EDA (17).
Lacrimation was improved in treated dogs except in one
treated at day 2 of age (Table 2). The correction of glands in
trachea, bronchi, and esophagus appeared, however, to be
dependent on the age at which the dogs were treated, i.e.
treatment administered earlier in life had a bigger impact on
gland development (Fig. 8 and Table 2). It is noteworthy that,
regardless of the extent of the phenotypic reversion, none of
the treated dogs suffered from pneumonia or other airway
diseases that are common in untreated EDA-deficient dogs
or from dry eye condition (keratoconjunctivitis sicca) that
affect all XLHED dogs.
DISCUSSION
Deficiency in the TNF family ligand EDA leads to ectoder-
mal dysplasia, even if the receptor EDAR remains fully func-
tional. The development of EDAR agonists are thus of inter-
est for applications in the treatment of XLHED. EDAR is
relatively well conserved across species, with only 4 amino
acid differences between human and dog, 10 between human
and mouse, and 13 between human and chicken in the 154-
amino acid-long mature extracellular domain (supplemental
Fig. S7). To increase the likelihood of getting cross-reactive
antibodies, EDAR-deficient mice were used to generate
monoclonal antibodies. The approach proved successful as
13 of the 15 anti-EDAR antibodies analyzed recognized
EDAR from human, dog, rat, mouse, and chicken. These
antibodies are therefore likely to cross-react with EDARs of
all mammals and many other vertebrates. Anti-EDAR15 dif-
ferentiated human and dog from rat and mouse EDARs
whose primary sequence only diverge in CRD1 (supplemen-
tal Fig. S7), implying that part of its epitope is in CRD1.
Anti-EDAR9 recognized CRD1–3 of EDAR but none of the
cysteine-rich domains taken individually or in pairs, despite
the fact that these fragments were overlapping and supposed
to respect structural elements of the receptor. These results
indicate that EDAR may fold on itself to create a conforma-
tional epitope with regions that are distant in the primary
FIGURE 7. Anti-EDAR antibody reverts many ectodermal dysplasia phenotypes in EDA-deficient mice. Pregnant Tabby mice were treated intravenously
at days 13 and 20 (E13/E20) of gestation with anti-EDAR antibody 3 at 16 mg/kg. Offspring were analyzed at 6 months of age. Age-matched wild type and
EDA-deficient Tabby mice were similarly analyzed for comparison. A, tail phenotype. B, transversal sections of tail skin showing the presence of hair follicles,
stained with hematoxylin and eosin. C, starch iodine sweat tests. D, sections of foot pads showing the presence of glandular structures of sweat glands
(arrowheads), stained with hematoxylin and eosin. E, top view of the retro-auricular region showing the presence of hair behind the ears. F, sections of the
trachea stained with Alcian blue to reveal mucus-secreting glands (arrowheads). G, eye phenotype showing reversion of the thickened eyelid margin and
narrow eyelid opening. Hand I, pictures of the jaw carrying the upper and lower molars. Antibody treated and wild type jaws carry larger teeth with a normal
pattern of cusps on their surfaces.
Agonist Anti-EDAR Antibodies
30776 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 •NUMBER 35 •SEPTEMBER 2, 2011
by guest on January 13, 2016http://www.jbc.org/Downloaded from
sequence. In any case, results indicate that agonist anti-
EDAR antibodies can recognize at least three distinct
epitopes in EDAR.
Anti-EDAR antibodies were screened in newborn Tabby
mice, a highly relevant in vivo assay whose main limitation is to
detect only antibodies cross-reacting with mouse EDAR. More
than half of the 46 hybridoma supernatants tested were active in
this assay, indicating that agonist antibodies can readily be
obtained with the procedure used. The success rate of detection
was lower using the EDAR:Fas reporter cell lines that signal
cell death in an oligomerization-sensitive manner, probably
because these cell-based assays make use of a different intracel-
lular signaling pathway. When combined, these assays discrim-
inated two classes of agonist anti-EDAR antibodies, with or
without in vitro activity. Interestingly, the in vitro activity cor-
related relatively well with low antibody dissociation constants
but not with association constants or affinities (supplemental
Fig. S8). The EDAR:Fas reporter cells were previously shown
to discriminate recombinant WT EDA1 and EDA1 with the
V365A mutation identified in a family with selective tooth
agenesis, despite the fact that these two ligands bind EDAR-Fc
almost equally well (14). This led to the hypothesis that forma-
tion of teeth may require higher quality EDAR signals than
those needed for hair or gland formation. Thus, it is possible
that agonist antibodies with activity in vitro may also be the best
ones to correct tooth defects associated with EDA deficiency. It
will be interesting to experimentally test this hypothesis in the
future.
The plethora of anti-EDAR agonist antibodies obtained,
most of which were of the IgG1 isotype, was surprising.
Indeed, there are indications that agonist anti-Fas antibodies
(Fas is another TNFR family member) need to be oligomer-
ized to be active. For example, the CH11 IgM monoclonal
antibody directed against human Fas was obtained by immu-
nization of mice with membranes of FS-7 human fibroblasts
(27). When an IgG1 recognizing the exact same epitope
(mAb ZB4) or a divalent F(ab⬘)
2
of CH11 was used, there was
no agonist activity (28). A second example of an agonist
FIGURE 8. Anti-EDAR antibody ameliorates dentition and presence of glands in EDA-deficient dogs. An EDA-deficient dog was treated at day 2 of
life with a single dose of anti-EDAR (antibody 3) at 10 mg/kg, and analyzed 1.6 years later in comparison with a wild type and with an affected dog. Aand
B, front and side views of the jaws. Cand D, hematoxylin and eosin-stained tissue sections of the trachea and bronchi. Glandular tissues are indicated
with arrowheads.
TABLE 2
Summary of clinical and pathological findings in untreated XLHED dogs and XHLED dogs treated with anti-EDAR antibody 3
Dog number Treatment protocol
Age at
necropsy
Appearance of teeth
(number)
Tracheal
glands
Bronchial
glands
Esophageal
glands Tear production
%
Wild type (n⫽5) 1–3 years Normal (41.6 ⫾0.9) ⫹⫹⫹ ⫹⫹⫹ ⫹⫹⫹ 97 ⫾14
XLHED (n⫽6) 1–3 years Abnormal (18.0 ⫾2.9) None None None 68 ⫾20
E237 10 mg/kg on day 2 1.7 years Greatly improved (40) ⫹⫹⫹ ⫹⫹⫹ ⫹⫹⫹ 70.0 ⫾4.1
E241 10 mg/kg on day 2 1.6 years Greatly improved (41) ⫹⫹ ⫹ ⫹⫹ 90.0 ⫾7.1
E222 7 mg/kg on day 14 2.4 years Improved (30) ⫹⫺ ⫹ 80.0 ⫾27.1
Agonist Anti-EDAR Antibodies
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30777
by guest on January 13, 2016http://www.jbc.org/Downloaded from
monoclonal antibody directed against human Fas is APO-1,
which was obtained by immunizing mice with plasma mem-
branes of the human SKW 6.4 cell line (29). This antibody is
an IgG3. Upon isotype switch, it loses its agonist activity (30),
a result that was tentatively explained by the propensity of
IgG3 to self-aggregate. As immunoglobulin preparations
often contain low amounts of high molecular weight anti-
body aggregates (31), we wondered whether the agonist
activity of anti-EDAR antibodies could be due to aggregates.
This was not the case, however, as monomeric antibodies
had a similar specific activity in vivo as the total preparation.
In addition, a purified F(ab⬘)
2
fragment was agonist in vivo,
whereas the Fab fragment was not. We conclude that diva-
lency is necessary and sufficient for anti-EDAR agonist anti-
bodies to exert their activity.
We have shown previously that the collagen domain of
EDA oligomerizes the trimeric TNF homology domain of
EDA1 into higher order structures with concomitant gain
of activity (13). Similarly, Fc-EDA1 that assembles as a hexamer
(containing two EDA1 trimers) is a highly active molecule.
One interpretation is that multiple EDAR molecules must be
recruited within the same complex to deliver robust intra-
cellular signals, but this is in apparent contradiction with the
observation that divalent antibodies are good agonists. A
first hypothesis to reconcile these observations is that EDAR
may pre-exist as inactive complexes before ligand binding, as
shown previously for Fas (32). Binding of a ligand may
change the conformation of the complex to render it signal-
ing-competent and bring together two such complexes to
initiate signaling. Divalent agonist antibodies may mimic
hexavalent ligands by inducing the conformational change
by binding one receptor in the pre-assembled complex and
recruiting and activating a second complex on its second
arm. In a second hypothesis, binding of the ligand or the
antibody at an appropriate site of EDAR is sufficient to
render the receptor signaling-competent. Assembly of the
signaling complex may be relatively slow and reversible if
the agonist detaches prematurely from the receptor. In this
model, efficient signaling could be obtained either with
divalent reagents with low dissociation constants or with
ligands that compensate relatively high dissociation con-
stants by multivalency. Whatever their mechanism of action,
Fc-EDA1 and agonist anti-EDAR antibodies are in practice
excellent agonists to cure animal models of XLHED, in-
cluding their teeth defects. Because of their long half-life
in vivo, agonist anti-EDAR antibodies will prove useful
reagents for long term experiments, especially in mice where
these mouse antibodies should elicit minimal neutralizing
immune responses.
Finally, it is noteworthy that keratoconjunctivitis sicca (dry
eye) that affects all untreated EDA-deficient dogs is believed to
be caused by decreased tear production. Tear production
improved significantly in two of the treated dogs but not in a
third one (E237). Nevertheless, none of the dogs treated in this
study with agonist anti-EDAR antibody required therapy for
dry eye or any other ocular disorders. These findings suggest
that decreased tear production is not the only factor causing dry
eyes but that other structures such as Meibomian glands that
lubricate the eye, or the composition of the lipids therein, also
play an important role.
Acknowledgments—We thank Christophe Maier (Edimer Biotech,
Epalinges) for invaluable input in the creation of Edimer; Jeff Behrens
and Neil Kirby for their motivating discussions, support, and for the
gift of Fc-EDA; Ste´phane Germain for assistance in early phases of
mice work; Olivier Donze´ for expert help in the generation of the anti-
bodies; John Lewis for dental pictures of the dogs; veterinary techni-
cians and students of the University of Pennsylvania for the expert
care of the dogs; and Ju¨rg Tschopp for continuous support and helpful
discussions.
REFERENCES
1. Aggarwal, B. B. (2003) Nat. Rev. Immunol. 3, 745–756
2. Mikkola, M. L. (2008) Cytokine Growth Factor Rev. 19, 219 –230
3. Yan, M., Wang, L. C., Hymowitz, S. G., Schilbach, S., Lee, J., Goddard, A.,
de Vos, A. M., Gao, W. Q., and Dixit, V. M. (2000) Science 290, 523–527
4. Headon, D. J., Emmal, S. A., Ferguson, B. M., Tucker, A. S., Justice,
M. J., Sharpe, P. T., Zonana, J., and Overbeek, P. A. (2001) Nature 414,
913–916
5. Headon, D. J., and Overbeek, P. A. (1999) Nat. Genet. 22, 370 –374
6. Monreal, A. W., Zonana, J., and Ferguson, B. (1998) Am. J. Hum. Genet. 63,
380–389
7. Newton, K., French, D. M., Yan, M., Frantz, G. D., and Dixit, V. M. (2004)
Mol. Cell. Biol. 24, 1608–1613
8. Srivastava, A. K., Pispa, J., Hartung, A. J., Du, Y., Ezer, S., Jenks, T., Shi-
mada, T., Pekkanen, M., Mikkola, M. L., Ko, M. S., Thesleff, I., Kere, J., and
Schlessinger, D. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 13069–13074
9. Nakata, M., Koshiba, H., Eto, K., and Nance, W. E. (1980) Am. J. Hum.
Genet. 32, 908–919
10. Pinheiro, M., and Freire-Maia, N. (1979) Am. J. Med. Genet. 4, 113–122
11. Zankl, A., Addor, M. C., Cousin, P., Gaide, A. C., Gudinchet, F., and Schor-
deret, D. F. (2001) Eur. J. Pediatr. 160, 296–299
12. Schneider, P., Street, S. L., Gaide, O., Hertig, S., Tardivel, A., Tschopp, J.,
Runkel, L., Alevizopoulos, K., Ferguson, B. M., and Zonana, J. (2001) J. Biol.
Chem. 276, 18819–18827
13. Swee, L. K., Ingold-Salamin, K., Tardivel, A., Willen, L., Gaide, O., Favre,
M., Demotz, S., Mikkola, M., and Schneider, P. (2009) J. Biol. Chem. 284,
27567–27576
14. Mues, G., Tardivel, A., Willen, L., Kapadia, H., Seaman, R., Frazier-Bow-
ers, S., Schneider, P., and D’Souza, R. N. (2010) Eur. J. Hum. Genet. 18,
19–25
15. Mustonen, T., Pispa, J., Mikkola, M. L., Pummila, M., Kangas, A. T., Pak-
kasja¨rvi, L., Jaatinen, R., and Thesleff, I. (2003) Dev. Biol. 259, 123–136
16. Cui, C. Y., Durmowicz, M., Ottolenghi, C., Hashimoto, T., Griggs, B.,
Srivastava, A. K., and Schlessinger, D. (2003) Hum. Mol. Genet. 12,
2931–2940
17. Casal, M. L., Lewis, J. R., Mauldin, E. A., Tardivel, A., Ingold, K., Favre, M.,
Paradies, F., Demotz, S., Gaide, O., and Schneider, P. (2007) Am. J. Hum.
Genet. 81, 1050–1056
18. Gaide, O., and Schneider, P. (2003) Nat. Med. 9, 614 – 618
19. Casal, M. L., Scheidt, J. L., Rhodes, J. L., Henthorn, P. S., and Werner, P.
(2005) Mamm. Genome 16, 524–531
20. Bossen, C., Ingold, K., Tardivel, A., Bodmer, J. L., Gaide, O., Hertig, S.,
Ambrose, C., Tschopp, J., and Schneider, P. (2006) J. Biol. Chem. 281,
13964–13971
21. Schneider, P. (2000) Methods Enzymol. 322, 325–345
22. Wang, Z., Raifu, M., Howard, M., Smith, L., Hansen, D., Goldsby, R., and
Ratner, D. (2000) J. Immunol. Methods 233, 167–177
23. Schneider, P., Holler, N., Bodmer, J. L., Hahne, M., Frei, K., Fontana, A.,
and Tschopp, J. (1998) J. Exp. Med. 187, 1205–1213
24. Rawlins, E. L., and Hogan, B. L. (2005) Dev. Dyn. 233, 1378 –1385
25. Mauldin, E. A., Gaide, O., Schneider, P., and Casal, M. L. (2009) Am. J.
Med. Genet. A 149A, 2045–2049
Agonist Anti-EDAR Antibodies
30778 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 •NUMBER 35 •SEPTEMBER 2, 2011
by guest on January 13, 2016http://www.jbc.org/Downloaded from
26. Shawlot, W., Siciliano, M. J., Stallings, R. L., and Overbeek, P. A. (1989)
Mol. Biol. Med. 6, 299–307
27. Yonehara, S., Ishii, A., and Yonehara, M. (1989) J. Exp. Med. 169,
1747–1756
28. Fadeel, B., Thorpe, C. J., Yonehara, S., and Chiodi, F. (1997) Int. Immunol.
9, 201–209
29. Trauth, B. C., Klas, C., Peters, A. M., Matzku, S., Mo¨ller, P., Falk, W.,
Debatin, K. M., and Krammer, P. H. (1989) Science 245, 301–305
30. Dhein, J., Daniel, P. T., Trauth, B. C., Oehm, A., Mo¨ller, P., and Krammer,
P. H. (1992) J. Immunol. 149, 3166–3173
31. Ahrer, K., Buchacher, A., Iberer, G., Josic, D., and Jungbauer, A. (2003)
J. Chromatogr. A 1009, 89–96
32. Siegel, R. M., Frederiksen, J. K., Zacharias, D. A., Chan, F. K., Johnson, M.,
Lynch, D., Tsien, R. Y., and Lenardo, M. J. (2000) Science 288, 2354–2357
Agonist Anti-EDAR Antibodies
SEPTEMBER 2, 2011•VOLUME 286• NUMBER 35 JOURNAL OF BIOLOGICAL CHEMISTRY 30779
by guest on January 13, 2016http://www.jbc.org/Downloaded from
Kowalczyk et al. Supplemental Material: Agonist anti-EDAR antibodies Page 1
Online Supplemental Material
Supplemental Figure 1. List of plasmids used in the study.
Amino acids are indicated with the one letter code or, for large stretches of sequence, by
a written description. Signal (signal peptide of haemaglutinin: MAIIYLILLFTAVRG | );
Flag (DYKDDDDK); hEDA1, hEDA2 (SwissProt accession number Q92838); human,
dog, rat, mouse, chicken EDAR (SwissProt accession numbers Q9UNEO, E2RA80,
D3ZGP2, Q9R187 and Q5EFZ7); hFc (IgG1 encoded by GenBank accession number
BC018747 or aa105-330 of SwissProt accession number P01857); hFas (SwissProt
accession number P25445); Ig signal (Signal peptide of mouse Ig, heavy chain:
MNFGFSLIFLVLVLKGVQC | EVKLV). The " | " indicate predicted proteolytic
cleavage sites by signal peptidase. Sequences of interest were cloned into the mammalian
expression vector PCR3 (Invitrogen) or into the retroviral expression vector pMSCV
(Clonetech). Note that ps2765 and ps2783 were obtained by point mutations of the mouse
and human sequences, respectively, and therefore are rat and dog sequences at the amino
acid level only.
Plasmid Designation Protein encoded Vector Figure
ps515 EGFP Enhanced green fluorescent protein PCR3 3
ps1938 Fc-EDA1 Signal-hIgG1 (aa 245-470)-hEDA1 (aa 238-391) PCR3 3, 5, 6
ps1377
pMSCV-puro
Modified pMSCV-puro (Clonetech) with HindIII-BglII-EcoRI-
NotI-XhoI-HpaI-ApaI cloning sites
pMSCV
ps2199 hEDAR:Fas hEDAR (aa 1-183)-VD-hFas (aa 169-335) ps1377 5B
ps2260 mEDAR:Fas mEDAR (aa 1-183)-VD-hFas (aa 169-335) ps1377 5A
ps1235 Fc-EDA2 Signal-LD-hIgG1 (aa 245-470)-hEDA2 (aa 245-389) PCR3 3
ps1431 hEDAR-GPI hEDAR (aa 1-183)-VD-hTRAILR3 (aa 157-259) PCR3 3
ps2783 dogEDAR-GPI dogEDAR (aa 1-183)-VD-hTRAILR3 (aa 157-259) PCR3 3
ps2765 ratEDAR-GPI ratEDAR (aa 1-179)-VD-hTRAILR3 (aa 157-259) PCR3 3
ps1765 mEDAR-GPI mEDAR (aa 1-183)-VD-hTRAILR3 (aa 157-259) PCR3 3
ps2290 chEDAR-GPI chEDAR (aa 1-183)-VD-hTRAILR3 (aa 157-259) PCR3 3
ps548 Flag-EDA1 Signal-Flag-GPGQVQLQVD-mEDA1 (aa 245-391) PCR3 2
ps336 Flag-BAFF Signal-Flag-GPGQVQLQ-hBAFF (aa 137-285) PCR3 2
ps930 hEDAR-Fc hEDAR (aa 1-183)-VD-hIgG1 (aa 245-470) PCR3 2
ps2887 hEDAR (CRD1+2+3)-Fc hEDAR (aa 1-149)-VD-hIgG1 (aa 245-470) PCR3 2
ps2484 hEDAR (CDR1+2)-Fc hEDAR (aa 1-114)-VD-hIgG1 (aa 245-470) PCR3 2
ps2481 hEDAR(CRD1)-Fc hEDAR (aa 1-72)-VD-hIgG1 (aa 245-470) PCR3 2
ps2482 hEDAR (CRD2)-Fc Signal-LE-hEDAR (aa 71-114)-VD-hIgG1 (aa 245-470) PCR3 2
ps2524 hEDAR(CRD3)-Fc Signal-LE-hEDAR (aa 115-149)-VD-hIgG1 (aa 245-470) PCR3 2
ps2515 hEDAR(CRD2+3)-Fc Signal-LE-hEDAR (aa 71-149)-VD-hIgG1 (aa 245-470) PCR3 2
ps2508 hEDAR(CRD3+stalk)-Fc Signal-LE-hEDAR (aa 115-183)-VD-hIgG1 (aa 245-470) PCR3 2
ps2509 hEDAR(stalk)-Fc Signal-LE-hEDAR (aa 150-183)-VD-hIgG1 (aa 245-470) PCR3 2
ps815 mEDAR-Fc mEDAR (aa 1-183)-VD-hIgG1 (aa 245-470) PCR3 2
ps229 hFas-Fc hFas (aa1-170)-VDhIgG1 (aa 245-470) PCR3 2
Kowalczyk et al.
Supplemental Material: Agonist anti-EDAR antibodies Page 2
Supplemental Figure 2: Amino acid sequences of light and heavy chains variable
regions of anti-EDAR monoclonal antibodies.
Sequences start at the mature N-terminus. Complementarity determining regions (CDRs)
are highlighted in boxes. Putative junctions of the protein sections encoded by the V, D
and J genes, or by randomly added nucleotides (N) are indicated. The junction with the
constant region (C) is also shown. Note that the light chains of anti-EDAR antibodies 2
and 4 are identical, and the heavy chains of anti-EDAR antibodies 10 and 11 are
identical. Anti-EDAR antibodies 1, 3 and 8 have similar heavy and light chains, most
probably originating from the same VH and VL genes.
Kowalczyk et al.
Supplemental Material: Agonist anti-EDAR antibodies Page 3
Supplemental Figure 3: Half-life determination of agonistic anti-EDAR antibodies.
Wild type mice were intravenously injected with 200 µl of 1 mg/ml of anti-EDAR 1 or
anti-EDAR 3 antibodies. Serum samples were collected after 20 minutes, 1, 2, 8, 16 and
32 days. The concentration of the anti-EDAR mAb was determined by incubating serial
dilutions of serum in wells coated with human EDAR-Fc at 1 µg/ml, followed by
horseradish peroxidase-coupled anti-mouse IgG and OPD substrate. For analysis, the
serum dilutions giving OD = 1 (considered to represent the EC50) for each time points
were plotted as a function of time. An exponential curve was fitted on the series of points
except the time point 20 minutes. A half-lives of 10 to 11 days were thus determined for
anti-EDAR antibodies 1 and 3.
Kowalczyk et al.
Supplemental Material: Agonist anti-EDAR antibodies Page 4
Supplemental Figure 4: An Fab’
2 fragment of an agonist anti-EDAR antibody is active
in vivo, whereas a monomeric Fab fragment acts as an inhibitor.
A. Superdex-200 gel permeation chromatography elution profiles of ficin-generated Fab
and Fab’
2 fragments of anti-EDAR antibody 1, previously isolated by size exclusion
chromatography (see Fig. 4A). Main peaks were collected and used for in vivo
experiments.
B. The Fab’2 fragment was injected at 0.5 mg/kg in newborn Tabby mice. Tail hair
formation was recorded 3 weeks later. Administration of the Fab fragment at 4 mg/kg did
not induce tail hair formation (n=3).
C. The Fab’2 fragment was injected ip at 0.5 mg/kg in 3 days-old Tabby pups, which is
one of the latest time point at which tail hair formation can still be partially rescued on
the ventral side of the tail (on the left of the picture). The Fab fragment was injected at 4
mg/kg 4 h before administration of the Fab’2 fragment and at 6 mg/kg together with the
Fab’2 fragment. The Fab was then administered again at 10 mg/kg 24 and 48 h later. Co-
treatment with the Fab fragment prevented the action of the Fab’2 fragment.
Supplemental Figure 5: An anti-EDAR antibody reverts many ectodermal dysplasia
phenotypes in EDA-deficient mice.
A pregnant Tabby mouse was treated iv at day 9 and 17 (E9/E17) of gestation with anti-
EDAR 3 at 16 mg/kg. Offspring was analyzed at 6 months of age, as described in the
legend to Figure 7.
Kowalczyk et al.
Supplemental Material: Agonist anti-EDAR antibodies Page 5
Supplemental Figure 6: Long-term reversion of ectodermal dysplasia symptoms upon
treatment with agonist anti-EDAR antibodies.
A pregnant Tabby mouse was treated iv at day 13 (E13) of gestation with anti-EDAR 3 at
6.5 mg/kg. Offspring and age-matched Tabby mouse were analyzed at 26 months of age.
A. Tail. B. Tail tip, showing absence of a kink in the treated mouse. C. Back hair,
showing the presence of the longer guard hair in the treated mouse. D. Sweat test
showing the presence of functional sweat glands in the treated mouse (arrowhead). E.
Retro-auricular region. F. Eye.
Kowalczyk et al.
Supplemental Material: Agonist anti-EDAR antibodies Page 6
Supplemental Figure 7: Amino acid sequence of the mature extracellular domain of
EDAR in different species.
Divergent amino acids are highlighted with colours. The positions of the cystein-rich
domains (CRDs) and stalk are indicated.
Supplemental Figure 8: Correlation between low dissociation constants and agonist
activity of anti-EDAR antibodies in an in vitro reporter cell line assay.
Binding characteristics of the agonist anti-EDAR monoclonal antibodies (see Table 1)
were plotted against their activity score in vivo (1/EC50, see Table 1) and their activity
score in vitro. The activity score in vitro was determined as 8*(1/log(EC50 hEDAR)) +
5.6*(1/log(EC50 mEDAR)) (see Table 1 for EC50 values). The correction factors 8 and 5.6
were chosen so that the best antibody(ies) in the human or mouse EDAR:Fas assays
score(s) 8 points. Antibodies showing agonist activity in vitro are identified by their
numbers in the graphs.
Stéphane Demotz and Pascal Schneider
Favre, Douglas M. Jefferson, Denis J. Headon,
Giovanna Badic, Anne-Lise Etter, Manuel
Mauldin, Olivier Gaide, Aubry Tardivel,
Willen, Margret L. Casal, Elizabeth A.
Christine Kowalczyk, Nathalie Dunkel, Laure
Antibodies
Receptor (EDAR) Agonist Monoclonal
Characterization of Anti-ectodysplasin A
Molecular and Therapeutic
Developmental Biology:
doi: 10.1074/jbc.M111.267997 originally published online July 5, 2011
2011, 286:30769-30779.J. Biol. Chem.
10.1074/jbc.M111.267997Access the most updated version of this article at doi:
.JBC Affinity SitesFind articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted• When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2011/07/05/M111.267997.DC1.html
http://www.jbc.org/content/286/35/30769.full.html#ref-list-1
This article cites 32 references, 13 of which can be accessed free at
by guest on January 13, 2016http://www.jbc.org/Downloaded from