Content uploaded by Rosa Sorrentino
Author content
All content in this area was uploaded by Rosa Sorrentino on Nov 24, 2015
Content may be subject to copyright.
Available via license: CC BY 4.0
Content may be subject to copyright.
Allele-dependent Similarity between Viral and Self-peptide
Presentation by HLA-B27 Subtypes*
Received for publication, September 20, 2004, and in revised form, October 26, 2004
Published, JBC Papers in Press, November 10, 2004, DOI 10.1074/jbc.M410807200
Maria Teresa Fiorillo‡§, Christine Ru¨ ckert§¶, Martin Hu¨ lsmeyer§储, Rosa Sorrentino‡,
Wolfram Saenger储**, Andreas Ziegler¶, and Barbara Uchanska-Ziegler¶‡‡
From the ‡Dipartimento di Biologia Cellulare e dello Sviluppo, Universita` di Roma “La Sapienza,” via dei Sardi 70,
00185 Roma, Italy, the ¶Institut fu¨ r Immungenetik, Charite´-Universita¨ tsmedizin Berlin, Campus Virchow-Klinikum,
Humboldt-Universita¨ t zu Berlin, Spandauer Damm 130, 14050 Berlin, Germany, and the 储Institut fu¨r
Chemie/Kristallographie, Freie Universita¨ t Berlin, Takustrasse 6, 14195 Berlin, Germany
Molecular mimicry is discussed as a possible mecha-
nism that may contribute to the development of autoim-
mune diseases. It could also be involved in the differen-
tial association of the human major histocompatibility
subtypes HLA-B*2705 and HLA-B*2709 with ankylosing
spondylitis. These two subtypes differ only in residue
116 of the heavy chain (Asp in B*2705 and His in B*2709),
but the reason for the differential disease association is
not understood. Using x-ray crystallography, we show
here that the viral peptide pLMP2 (RRRWRRLTV, de-
rived from latent membrane protein 2 (residues 236 –
244) of Epstein-Barr virus) is presented by the B*2705
and B*2709 molecules in two drastically deviating con-
formations. Extensive structural similarity between
pLMP2 and the self-peptide pVIPR (RRKWRRWHL, de-
rived from vasoactive intestinal peptide type 1 receptor
(residues 400 –408)) is observed only when the peptides
are presented by B*2705 because of a salt bridge be-
tween Arg
5
of both peptides and the subtype-specific
heavy chain residue Asp
116
. Combined with functional
studies using pLMP2/pVIPR-cross-reactive cytotoxic T
cell lines and clones, together with target cells present-
ing these peptides or a modified peptide analogue, our
results reveal that a pathogen-derived peptide can ex-
hibit major histocompatibility complex class I subtype-
dependent, drastically distinct binding modes. Further-
more, the results demonstrate that molecular mimicry
between pLMP2 and pVIPR in the HLA-B27 context is an
allele-dependent property.
Not all HLA-B27 subtypes are equally associated with the
autoimmune disease ankylosing spondylitis (AS).
1
The fre-
quent, prototypical subtype B*2705 is AS-associated, inde-
pendent of ethnic origin, whereas B*2706 and B*2709, which
exhibit geographically restricted distribution, are not (1). The
products of the B*2705 and B*2709 alleles differ only in residue
116 of the HLA-B27 heavy chain (HC; Asp in B*2705 and His
in B*2709) (2). This residue is located at the floor of the pep-
tide-binding groove, forms part of the F-pocket, and is buried
upon binding of a peptide. Despite the close structural similar-
ities, the subtypes give rise to distinct repertoires of bound
peptides (3) and cytotoxic T lymphocytes (CTL) (4). B*2705-
positive but not B*2709-positive individuals possess CTL that
recognize the HLA-B27-bound self-peptide pVIPR (RRK-
WRRWHL). pVIPR is derived from vasoactive intestinal pep-
tide type 1 receptor (residues 400–408) and is presented by
B*2709 molecules in the common canonical conformation,
whereas B*2705 presents the peptide in an unusual dual bind-
ing mode (5).
pVIPR exhibits sequence homology with the peptide pLMP2
(RRRWRRLTV), derived from latent membrane protein 2 (res-
idues 236–244) of Epstein-Barr virus (EBV). The existence of
CTL reacting with both peptides in the context of B*2705
suggests a relationship between infection with EBV and an
expansion of the pool of pLMP2/pVIPR-cross-reactive CTL (4).
However, a direct correlation between EBV infection and AS
pathogenesis has not been established. Molecular mimicry (6–
9), i.e. similarity in overall shape as well as charge distribution
for an interaction surface (9), has been invoked as an explana-
tion for the association of HLA-B27 and spondyloarthropathies
(10–12), but its existence has yet to be proven (13, 14). A
principal difficulty is related to the fact that a host-derived
epitope is expected to share antigenic but not necessarily also
extensive sequence homology with a foreign antigen (15).
We have now determined the structures of both HLA-B27
subtypes in complex with pLMP2 and compare them here with
the corresponding pVIPR complexes. Together with functional
data, this comparison suggests that structural similarity and
CTL cross-reactivity between pLMP2 and pVIPR in the context
of HLA-B27 antigens are allele-dependent properties.
EXPERIMENTAL PROCEDURES
HLA-B27-positive Donors, CTL Lines, and Clones—Seven patients
with AS (with the exception of one B*2702-positive individual, all typed
as B*2705) and two healthy individuals (one B*2705-positive and one
B*2709-positive) were enrolled for this study (see Tables III, IV, and V).
HLA-B27 typing and generation of pLMP2- and pVIPR-specific CTL
lines were carried out as described (4). The CTL line MP VPAC7 was
cloned by limiting dilution at 0.5–1 cell/well in 96-well U-bottom micro-
plates in the presence of phytohemagglutinin (0.5
g/ml), 3 ⫻10
4
allogeneic
␥
-irradiated peripheral blood mononuclear cells, and 20
units/ml recombinant interleukin-2 (Roche Applied Science). After 12
days, the growing cells were restimulated with pVIPR-pulsed,
␥
-irradi-
* This work was financially supported by Deutsche Forschungsge-
meinschaft Grant SFB 449, TP B5,B6; Sonnenfeld-Stiftung; COFIN
2001; and Istituto Pasteur Fondazione Cenci-Bolognetti. The costs of
publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked “advertisement”
in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The atomic coordinates and structure factors (code 1uxs and 1uxw)
have been deposited in the Protein Data Bank, Research Collaboratory
for Structural Bioinformatics, Rutgers University, New Brunswick, NJ
(http://www.rcsb.org/).
§ These authors contributed equally to this work.
** To whom correspondence may be addressed. Tel.: 49-30-8385-
3412; Fax: 49-30-8385-6702; E-mail: saenger@chemie.fu-berlin.de.
‡‡ To whom correspondence may be addressed. Tel.: 49-30-4505-
53517; Fax: 49-30-4505-53953; E-mail: barbara.uchanska-ziegler@
charite.de.
1
The abbreviations used are: AS, ankylosing spondylitis; EBV, Ep-
stein-Barr virus; HLA, human leukocyte antigen; MHC, major histo-
compatibility complex; HC, heavy chain; CTL, cytotoxic T lympho-
cyte(s); HIV, human immunodeficiency virus; TCR, T cell receptor.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 280, No. 4, Issue of January 28, pp. 2962–2971, 2005
© 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org2962
ated autologous B-LCL and further expanded in the presence of recom-
binant interleukin-2 (20–50 units/ml).
Cytotoxicity Assays—Cytolytic activity of pLMP2-responsive
(RRRWRRLTV), pVIPR-responsive (RRKWRRWHL), and pVIPR-
pArg
3
-responsive (RRRWRRWHL) CTL lines and clones was assessed
according to the standard
51
Cr release procedure (4). Target cells
(B*2705 or B*2709 T2 transfectants) were incubated overnight with the
pLMP2 or pVIPR peptides at 70
Mor at lower concentrations (see
“Results” and Fig. 4 for details) or in medium alone. One day later, the
cells were labeled with sodium chromate and extensively washed before
being mixed with effector T cells at 3 ⫻10
3
target cells/well.
Analysis of TCR Gene Usage—Total RNA was extracted from 2 ⫻10
5
T cells, and cDNA was synthesized using oligo(dT) primer and Super-
Script
TM
II RNase H
⫺
Reverse Transcriptase (Invitrogen) according to
the manufacturer’s instructions. For the analysis of TCR
␣
chain usage,
cDNA was amplified using primers and PCR conditions as already
described (16). Additional oligonucleotides for V
␣
families 18–29 were
designed as detailed previously (17). PCR products were loaded on a
1.5% agarose gel stained with ethidium bromide. Specific DNA bands
were cut from the gel and purified using a gel band purification kit
(Amersham Biosciences). Direct sequencing was performed using an
internal primer upstream of the TCR C
␣
reverse primer.
Protein Preparation and Structure Determination—The peptide
pLMP2 was purified by high pressure liquid chromatography (Alta
Bioscience), and HLA-B27䡠peptide complexes were produced as de-
scribed (5). Purified complexes (15–20 mg/ml in 20 mMTris/HCl, 150
mMNaCl, 0.01% sodium azide, pH 7.5) were used for crystallization
using hanging drop vapor diffusion and streak-seeding. Crystals suit-
able for x-ray diffraction experiments were grown in drops made of 1.5
l of protein solution and 1.5
l of precipitant solution (for B*2705, 15%
polyethylene glycol 8000, 0.1 MTris/HCl, pH 7.5, and for B*2709, 21%
polyethylene glycol 8000, 0.1 MTris/HCl, pH 8.5). Using glycerol as
cryoprotectant, data sets were obtained from cryo-cooled (100 K) crys-
tals at the BL2 beam line of BESSY-II. The data were processed with
the HKL package (see Table I) (18).
The structure of B*2709䡠pLMP2 was determined by molecular re-
placement using peptide-stripped B*2709䡠m9 (Protein Data Bank code
1k5n) as a search model and the program Molrep (19). After rigid body
refinement using Refmac (20), the initial model was subjected to sim-
ulated annealing and energy minimization using CNS (21) to remove
model bias. Further refinement was carried out by iterative cycles of
manual rebuilding using O (22) and restrained maximum-likelihood
with Refmac comprising B-factor adjustment. Water molecules were
included with ARP/wARP (23). After translation, libration, screw rota-
tion refinement (24), the Rfactor converged at 0.154 (R
free
⫽0.190). As
the two HLA-B27䡠pLMP2 complexes crystallized isomorphously, initial
phases for B*2705䡠pLMP2 were calculated from peptide-stripped
B*2709䡠pLMP2 with His
116
replaced by alanine. This initial model was
subjected to rigid body refinement, simulated annealing, and energy
minimization using CNS, improved by manual intervention using O
and water molecule inclusion as described for the B*2709-data process-
ing. Because of the higher resolution of B*2705䡠pLMP2, restrained
maximum-likelihood refinement (Refmac) included anisotropic B-factor
refinement. Evaluation of the atomic displacement parameters by Par-
vati (25) provided the expected statistical distribution of 0.5 ⫾0.17 for
all atoms of the structure. Both structures were validated with What-
check (26), and the statistics are compiled in Table I. The figures were
generated using Delphi (27), Povray (www.povray.org), Molscript (28),
Rastop (www.geneinfinity.org/rastop/), MSMS (29), and Raster3D (30)
together with a graphical interface (Moldraw) developed by N. Stra¨ ter
(Institut fu¨ r Kristallographie, Freie Universita¨t Berlin).
2
The atomic coordinates and structure amplitudes have been depos-
ited in the Protein Data Bank under accession codes 1uxs
(B*2705䡠pLMP2) and 1uxw (B*2709䡠pLMP2).
RESULTS
Structural Features of pLMP2 in Complex with HLA-B27
Subtypes—The B*2705䡠pLMP2 and B*2709䡠pLMP2 complexes
crystallized isomorphously in space group P2
1
(Table I). Both
show the typical MHC class I topography (31) (Fig. 1, a– c) and
were refined at high resolution: 1.55 Å for B*2705䡠pLMP2 and
1.72 Å for B*2709䡠pLMP2. The HLA-B27 HC and

2
-micro-
globulin are highly similar in the two subtypes (except for the
D116H exchange) with a C
␣
root mean square deviation of only
0.2 Å. For each complex, the peptide could be modeled unam-
biguously to the electron density (Fig. 1, dand e). When com-
plexed to B*2709, pLMP2 is bound in the conventional p4
␣
conformation (main chain
/
torsion angles in
␣
-helical con-
formation at p4), with the solvent-exposed pArg
5
side chain
pointing away from the binding groove (Fig. 1b) (5). In contrast,
pLMP2 displays the drastically different p6
␣
conformation
(main chain
/
torsion angles in
␣
-helical conformation at p6)
when bound to B*2705 (Fig. 1a), with the side chain of pArg
5
pointing toward the interior of the binding groove, where it
forms a salt bridge with HC Asp
116
(5). These subtype-depend-
ent pArg
5
orientations force the middle portion (residues p4–
p7) of the peptide backbones and the corresponding amino acid
side chains into grossly different conformations in the two
2
N. Stra¨ ter, unpublished program.
TABLE I
Data collection and refinement statistics
HLA-B*2705䡠pLMP2 HLA-B*2709䡠pLMP2
Data collection
Space group P2
1
P2
1
Unit cell (Å, Å, Å; °) 51.1, 82.3, 65.9; 109.3 50.9, 82.6, 62.8; 104.4
Resolution (Å)
a
40.0–1.55 (1.61–1.55) 40.0–1.72 (1.78–1.72)
Unique reflections 74,438 (7225) 53,353 (5036)
Completeness (%)
a
99.4 (97.0) 99.4 (96.0)
I/
a
21.0 (4.1) 20.2 (3.7)
R
syma,b
0.051 (0.246) 0.045 (0.262)
Refinement
Nonhydrogen atoms 3992 3876
R
crysta,c
0.142 (0.179) 0.154 (0.183)
R
freea,d
0.177 (0.202) 0.190 (0.224)
Heavy chain, no. of atoms/average B factor (Å
2
)2301/20.0 2298/12.85

2
-Microglobulin, no. of atoms/average B factor (Å
2
)855/23.0 860/17.9
Peptide, no. of atoms/average B factor (Å
2
)102/19.6 92/10.3
Water, no. of molecules/average B factor (Å
2
)710/38.7 596/33.7
Glycerol, no. of atoms/average B factor (Å
2
)24/37.4 30/21.5
Estimated overall coordinate error (Å)
e
Root mean square deviation from ideal geometry
Bond length (Å) 0.015 0.015
Bond angles (°) 1.5 1.6
a
The values in parentheses refer to the highest resolution shell.
b
R
sym
⫽⌺
h
⌺
i
兩I
h,i
⫺具I
h
典兩/⌺
h
⌺
i
I
h,i
.
c
R
cryst
⫽⌺
h
兩F
o
⫺F
c
兩/⌺F
o
(working set, no
cut-off applied).
d
R
free
is the same as R
cryst
, but calculated on 5% of the data excluded from refinement.
e
Estimated overall coordinate error based on R
free
as calculated by Refmac 5.1.1999.
Molecular Mimicry in HLA-B27 Subtypes 2963
subtypes (Fig. 1c). Because these regions of the two complexes
do not participate in extensive crystal contacts, the observed
conformational differences must be a direct consequence of the
D116H polymorphism.
The N-terminal pLMP2 residues pArg
1
, pArg
2
, and pArg
3
oc-
cupy identical positions in both subtypes (Fig. 1cand Table II).
Both subtypes exhibit also closely related interactions between
HC atoms and C-terminal peptide residues pThr
8
and pVal
9
.In
B*2709, however, p8 and p9 are located slightly deeper in the
binding groove than in B*2705 (Fig. 1, a– c), possibly as a conse-
quence of altered p4–p7 conformations. To account for these
changes, the binding groove residues in contact with pThr
8
and
pVal
9
(Table II) exhibit small side chain variations.
pTrp
4
is the first pLMP2 residue with substantially different
positioning in the two subtypes (Fig. 1, a– e, and Table II). In
p4
␣
conformation found in complex with B*2709, the pTrp
4
,
pArg
5
, and pArg
6
side chains are fully solvent-exposed, with
few HC contacts, whereas pLeu
7
projects into the E-pocket
(Fig. 1, b,c, and e, and Table II). A very different situation is
found in the p6
␣
conformation seen in B*2705 (Fig. 1, a,c, and
d). Here, the pTrp
4
side chain is packed against the
␣
1-helix,
and pArg
5
forms a salt bridge with Asp
116
that leads to deeper
insertion of the middle section of the peptide into the binding
groove (Fig. 1aand Table II). At pArg
6
, the peptide backbone
bends upward (associated with the p6
␣
conformation), so that
the side chain can engage in van der Waals’ contact with pTrp
4
(Table II). Finally, pLeu
7
is solvent-exposed in B*2705, and its
side chain exhibits the highest flexibility of all pLMP2 residues
in either conformation as shown by temperature (B) factors
(Fig. 1, fand g, and Table I). A comparison of the B factors of
the peptide reveals that pLMP2 is more flexibly bound in
B*2705 despite the anchoring of its middle through the pArg
5
-
Asp
116
interaction. This differential peptide flexibility is most
likely a consequence of a network of solvent molecules that is
tighter in B*2709 than in B*2705, where the hydrophobic sec-
tion of the pArg
5
side chain prevents its formation.
Structural Comparison of pLMP2 and pVIPR in Complexes
with B*2705 and B*2709 —The four complexes of B*2705 and
B*2709 with pLMP2 and pVIPR, respectively, crystallized
isomorphously (Ref. 5 and Table I), indicating that the same
crystallographic restraints (intermolecular interactions asso-
ciated with crystal packing) apply to all of them. Comparison
of pVIPR complexed with B*2705 and with B*2709 has al-
ready been carried out, with the dual p4
␣
/p6
␣
conformation
found in B*2705 but not in B*2709, where only p4
␣
occurs (5).
Therefore, if molecular mimicry were to play a role in the
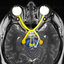
FIG.1.pLMP2 topographies, elec-
tron densities, and B factors when
bound to B*2705 and B*2709. aand b,
conformation of pLMP2 in B*2705 and
B*2709, viewed from the side of the
␣
2-
helix together with a ribbon representa-
tion of the
␣
1-helix and the floor (

-sheet)
of the binding groove. The subtype-spe-
cific residue 116 is indicated (Asp
116
,red;
His
116
,blue), with the bidentate salt
bridge to Asp
116
shown as dotted lines.In
b, pArg
5
points toward the viewer. c,su
-
perimposition of pLMP2 in B*2705 and
B*2709, viewed as in aand b, showing
that the peptide conformations in the two
subtypes differ drastically from pTrp
4
to
pThr
8
.dand e, final 2F
o
⫺F
c
electron
density contoured at the 1
level of
pLMP2 conformations in B*2705 (d) and
B*2709 (e). Water molecules are omitted
for clarity from the representations in
a– e.fand g, pLMP2 bound by B*2705 (f)
and B*2709 (g), color-coded by isotropic B
factor. A quantitative comparison is made
difficult by the refinement strategies em-
ployed (anisotropic for B*2705 and isotro-
pic for B*2709).
Molecular Mimicry in HLA-B27 Subtypes2964
context of the pLMP2/pVIPR structures, as suggested by CTL
cross-reactivity (4), a comparison of side chain orientations
(Fig. 2) and surface properties (Fig. 3) between the two
pLMP2 complexes and the two pVIPR complexes should pro-
vide a structure-based explanation.
The structures of the two peptides in B*2705 immediately
reveal that pLMP2 (Fig. 3a) is much more similar to pVIPR-
p6
␣
(C
␣
root mean square deviation of 0.3 Å; Fig. 3c) than to
pVIPR-p4
␣
(C
␣
root mean square deviation of 1.6 Å; Fig. 3d).
pArg
1
as well as the anchor residues pArg
2
and pVal
9
/pLeu
9
occupy virtually identical positions (Fig. 2, aand b). As ex-
pected from the peptide sequences, the similarity of pVIPR and
pLMP2 is most pronounced in the N-terminal half, extending to
pArg
6
. In addition to the amino acid exchange at p7, the sol-
vent-exposed pHis
8
in pVIPR and pThr
8
in pLMP2 lead to a
marked topographical change near the peptide C termini (Figs.
2, aand b, and 3, aand c). The similarity between
B*2705䡠pLMP2 (p6
␣
binding mode) and B*2705䡠pVIPR-p6
␣
ex-
tends beyond conformational (Figs. 2, aand b, and 3, aand c)
to electrostatic properties of their surfaces (Fig. 3, eand g),
particularly in the N-terminal halves of the peptides. In con-
trast, pLMP2 and pVIPR-p4
␣
in B*2705 are less equivalent at
residues p4–p8 (Figs. 2, cand d, and 3, aand d). The electro-
static surfaces of the two complexes in p4
␣
/p6
␣
conformation
diverge considerably as well (Fig. 3, eand h). It seems therefore
plausible to conclude that pLMP2/pVIPR CTL cross-reactivity
is more likely to occur when pVIPR is displayed in p6
␣
than in
p4
␣
conformation.
Surprisingly, the p4
␣
conformations of the two peptides
found in B*2709 differ much more (C
␣
root mean square devi-
ation of 0.9 Å; Figs. 2, eand f, and 3, b,d,f, and h) than the two
p6
␣
conformations in B*2705 (Figs. 2, aand b, and 3, a,c,e, and
g). Again, residues p1, p2, p4, and p9 show negligible varia-
tions, and pLys
3
(pVIPR) also occupies a similar position as
pArg
3
(pLMP2). However, the side chain guanidinium moieties
of both pArg
5
residues are solvent-exposed and point to oppo-
site directions (Figs. 2, eand f, and 3, band d), whereas those
in p6
␣
conformation (Figs. 2, aand b, and 3, aand c) are buried.
pArg
6
displays a substantial difference as well; in pLMP2, this
side chain is completely solvent-accessible with only a few
contacts to the
␣
1-helix, whereas it wedges between the peptide
backbone and the
␣
1-helix in the complex with pVIPR (5). The
C
␣
atoms of pArg
6
deviate by 2.3 Å, and the disparity between
the guanidinium groups is even larger. Although more simi-
larly positioned, the p7-C
␣
atoms still differ by 1.6 Å. The side
chain of pTrp
7
(pVIPR) occupies the front part of the large
E-pocket, an impossible location for pLeu
7
(pLMP2) because of
steric hindrance exerted by the large pArg
3
located in the
neighboring D-pocket. As a consequence, pLeu
7
occupies the
back part of the E-pocket, toward the peptide C terminus, is
inserted deeper than pTrp
7
in pVIPR, and is shifted toward the
␣
1-helix. The peptides deviate only by 1.1 Å at C
␣
of p8, but the
TABLE II
Comparison of pLMP2 peptide coordination in the B*2705 and B*2709 subtypes
Only direct intrapeptide contacts and contacts between pLMP2 and HC residues are included, and solvent-mediated interactions are omitted.
van der Waals’ contacts are not given explicitly for each amino acid. In the B*2705 subtype, pTrp
4
and Asp
116
occur in alternative conformations.
Only one of the equally occupied pTrp
4
conformations and the higher occupied Asp
116
conformation (q⫽0.75) are shown and discussed in the text.
a
Intrapeptide contact.
b
Helix
␣
1.
c

-Sheet floor.
d
Helix
␣
2.
Molecular Mimicry in HLA-B27 Subtypes 2965
side chains display different shapes (Fig. 3, band d) and
electrostatic potentials (Fig. 3, fand h). These similarities and
differences suggest that pLMP2/pVIPR-cross-reactive CTL
from B*2709 individuals are likely to recognize predominantly
epitopes formed by p1–p4 (Fig. 2g) and the adjacent residues of
the binding groove.
Peptide- and Subtype-dependent CTL Recognition—We have
previously shown that autoreactive CTL lines from individuals
typing as B*2705 are frequently observed in patients with AS
but occur in reduced numbers in healthy individuals. Such CTL
are only rarely found in B*2709 individuals, and pLMP2/
pVIPR-cross-reactive CTL are infrequently observed among
pLMP2-reactive CTL from either subtype (4). To better under-
stand the nature of this cross-reactivity, we have now carried
out more extensive studies with CTL, also including CTL
clones as well as a hybrid peptide (pVIPR-pArg
3
) in which
pLys
3
(from pVIPR) is replaced by pArg
3
(as in pLMP2; Tables
III, IV, and V). This exchange was thought to influence epitope
recognition by cross-reactive CTL. The ability of pVIPR-pArg
3
to stabilize both B*2705 and B*2709 molecules has been eval-
uated as already described for pVIPR and pLMP2 (Ref. 4 and
data not shown).
pLMP2-stimulated CTL from either HLA-B27 subtype detect
this peptide also in the context of the other subtype (Table III),
implying that structurally similar regions around p1–p3 or p8
provide the epitope(s) (Fig. 1c). About one-sixth of these CTL
cross-react with pVIPR at high peptide concentration (70
M),
irrespective of the subtype presenting this peptide. Further-
more, about 25% of the pVIPR-stimulated CTL from B*2705
individuals cross-react with pLMP2 and recognize both pep-
tides also in the B*2709 context. In addition, all pVIPR-stim-
ulated CTL from B*2705 individuals that cross-react with
pLMP2 recognize both peptides also in the B*2709 subtype
(Table IV). Four pLMP2/pVIPR-cross-reactive CTL lines, two
from a B*2709-positive individual (Ci) and two from a B*2705-
positive AS patient (MP), were also tested at lower peptide
concentrations (Fig. 4). It is evident that when B*2709 presents
the two peptides, the dose-response curves have a similar pro-
file (Fig. 4, b,d,f, and h). In contrast, when it is B*2705 that
displays the two peptides, the CTL of the patient show a strik-
ingly higher preference for pLMP2 (Fig. 4, eand g), even
though they derive from a stimulation with pVIPR. In case of
B*2705, the difference between the two peptides was found to
be about 100-fold.
The reactivity against pVIPR and pLMP2 either in the
B*2705 or in the B*2709 context has been tested also with
several clones derived from the CTL line MP VPAC7. Repre-
sentative dose-response curves of one CTL clone are shown
using also lower peptide concentrations (Fig. 4, iand j). The
profiles are similar to those obtained with the parental CTL
line; in particular, we found that the clonal reactivities were
always much stronger against pLMP2, although this peptide
was not employed for the initial stimulations of the CTL. TCR
gene usage was assessed for 17 clones derived from MP VPAC7.
The results obtained with clone 8 (Fig. 4, iand j) are represent-
ative for the others. The same TCR-AV14 chain, CDR3 motif
(DRDDKI), and J segment (AJ9S4) were found (Fig. 4k). A
higher degree of variability was found for TCR

-chains, but
TCRBV1BJ2S1 chains were predominant among the MP
VPAC7-derived clones.
Experiments with the pVIPR-pArg
3
peptide show that even a
conservative amino acid replacement in the N-terminal half of
FIG.2. Comparison of pVIPR and
pLMP2 as presented by B*2705 and
B*2709. Superimpositions in two views
rotated 90° with respect to each other;
selected amino acids are indicated. aand
b, superimposition of pLMP2 (yellow) and
pVIPR (magenta), both in p6
␣
binding
mode, as presented by B*2705 molecules,
viewed from the side (a, same view as in
Fig. 1a) or the top (b). cand d, superim-
position of pLMP2 (yellow,p6
␣
conforma-
tion) and pVIPR-p4
␣
(blue) as presented
by B*2705 molecules, viewed from the
side (c) or the top (d). eand f, superimpo-
sition of pLMP2 (green) and pVIPR (blue),
both in p4
␣
binding mode, as presented by
B*2709 molecules, viewed from the side
(e) or the top (f). g,left panel, schematic
description of side chain orientations
when looking from the N to the C termini
of pLMP2, pVIPR-p6
␣
, and pVIPR-p4
␣
in
the two HLA-B27 subtypes. The shaded
areas indicate structural similarity be-
tween the two peptides as presented by
B*2705 (dark gray) and B*2709 (light
gray). Right panel, floor of peptide bind-
ing groove indicated by

-sheet, and bind-
ing region for TCR indicated by TCR.
Molecular Mimicry in HLA-B27 Subtypes2966
the pVIPR peptide (pLys
3
Arg) 32 can lead to considerable al-
terations in the reactivity of CTL lines (Table V). pVIPR-pArg
3
abolishes or diminishes the reactivity of the majority of the
pVIPR-specific CTL. Lack of reactivity is unlikely to be influ-
enced by the fact that oligoclonal CTL lines were used in the
studies reported here, because their oligoclonality would be
expected to enhance and not to diminish their cross-reactive
potential. Only two CTL (AB4 and AB5) were found that re-
FIG.3. Molecular surfaces of
pLMP2 and pVIPR as presented by
B*2705 and B*2709. Molecular surface
representations of B*2705 (a,c,e, and g)
and B*2709 (b,d,f, and h) complexed
with pLMP2 or pVIPR, as viewed by an
approaching TCR; color-coding is as in
Figs. 1 and 2. a– d demonstrate shape
similarities and differences, whereas e– h
show the electrostatic surfaces. pVIPR-
p4
␣
is presented identically by B*2705
and B*2709 (dand h) (5). Red indicates a
negative surface charge, blue indicates a
positive surface charge, and gray areas
are uncharged.
TABLE III
pLMP2-stimulated CTL and assessment of pLMP2/pVIPR cross-reactivity
Donor
a
Tested on
B*2705䡠pLMP2 Tested on
B*2709䡠pLMP2 Tested on
B*2705䡠pVIPR Tested on
B*2709䡠pVIPR
CV 4/4
b
4/4 0/4 0/4
MP 4/4 4/4 2/4 2/4
MA 3/3 3/3 0/3 0/3
BO 1/1 1/1 0/1 0/1
EP 4/4 4/4 0/4 0/4
Ci 9/9 9/9 1/4, 5 NT
c
2/9
a
Donor CV is a healthy HLA-B*2705-positive individual. Donors MP, MA, BO, and EP are HLA-B*2705-positive patients with AS, and donor
Ci is an HLA-B*2709-positive healthy individual.
b
No. of CTL with positive reactivity/total no. of CTL tested.
c
NT, not tested.
Molecular Mimicry in HLA-B27 Subtypes 2967
tained their level of pVIPR-directed reactivity also with pVIPR-
pArg
3
(B*2709 not tested), and two others (PM53 and LV1) had
reduced activity (B*2702-derived LV1 not tested on B*2709). A
single CTL (PM 63) reacted with pVIPR-pArg
3
only in the
context of B*2709, although pVIPR was recognized when pre-
sented by both subtypes.
DISCUSSION
The results presented here show that a pathogen-derived
peptide such as pLMP2 can exhibit MHC class I subtype-de-
pendent, drastically distinct binding modes (Fig. 1). Although
pLMP2 is not an immunodominant peptide for HLA-B*2705
(32), specific CTL are readily detectable in individuals with this
subtype (4), and cross-reactivity between B*2705 and B*2709
presenting this peptide is invariably observed (Tables III, IV,
and V), most likely because of the existence of nearly identical
structures around the N- or C-terminal regions of the com-
plexes (p1–p3 and p8–p9; Figs. 2 and 3 and Table II). Under
natural conditions, CTL cross-reactivity between the subtypes
will be experienced only very rarely; because the B*2709 sub-
type is found exclusively among Sardinians (1, 2), one can
calculate that there are currently only about 100 living B*2705/
B*2709 heterozygous individuals (the frequency of the two
subtypes in Sardinia is about 1.6 and 0.4%, respectively (2, 33).
On the other hand, the worldwide distribution of the B*2705
allele (1) and the very frequent development of immune re-
sponses against EBV (34, 35) indicate that cross-reactivity
between the pLMP2 and pVIPR peptides in the context of
B*2705 might be much more frequent.
Do the structural findings obtained at 100 K correlate with
pLMP2/pVIPR CTL cross-reactivity assessed at 37 °C in the
context of HLA-B27? It is principally possible that pLMP2 and
also pVIPR exhibit conformations at physiological temperature
that are not identical to those found in the crystals grown at
18 °C. However, we regard this as extremely unlikely because
until now no case has been reported to our knowledge where a
crystal structure suffered significant changes when diffraction
data were collected at ambient and 100 K temperatures (36,
37). Similarly, the folding of the polypeptide chains in protein
structures determined in solution by NMR and in the crystal-
line state by x-ray diffraction are comparable as are the con-
formations of side chains in the protein interior (38–40). Dif-
ferences have been observed with side chains at the periphery
that are engaged in crystal contacts, and significant differences
have been reported in selected cases, e.g. for the inflammatory
protein C3a and interleukin 8 (38). The issue of structural
identity or similarity at different temperatures and states
could eventually be resolved with dynamical studies of these
molecules at physiological temperature (41) that must be con-
ducted in vitro. Consequently, purists could again raise ques-
tions concerning the behavior in vivo. We have, however, taken
great care that the peptides were present during the cytotox-
icity tests only in HLA-B27-bound form by washing the target
cells before exposure to CTL. This should favor the exclusive
presence of the thermodynamically most advantageous confor-
mation, which is very probably that or very close to that ob-
served in the crystals.
Because the peptides exhibit sequence dissimilarity from p7
to p9 (neglecting the conservative exchange pArg
3
/pLys
3
) asso-
ciated with differences in shape and charge distribution (Fig.
3), it seems likely that the N-terminal halves of the peptides
give rise to functional and structural mimicry. Remarkably,
only one of the two pVIPR conformations (p6
␣
) shows extensive
structural mimicry with pLMP2 (Figs. 2 and 3). Here, struc-
tural equivalence extends at least from residues p1 to p6 and
might even include the area above residue p7. Functional mo-
lecular mimicry in B*2705, i.e. CTL cross-reactivity between
pLMP2 and pVIPR, could be facilitated also by the higher
flexibility exhibited by pLMP2 when bound to this subtype as
compared with B*2709 (Fig. 1).
In contrast, cross-reactivity between the conformations of
pLMP2 (p6
␣
binding mode) and pVIPR-p4
␣
seems much less
likely (Figs. 2 and 3). Not only the overall shapes of the two
structures are distinct, but the electrostatic surfaces exhibit
considerable dissimilarity as well. Only the surfaces around
TABLE IV
pVIPR-stimulated CTL and assessment of pVIPR/pLMP2 cross-reactivity (only pVIPR-stimulated, pLMP2-cross-reactive CTL are shown)
CTL
a
Tested on
B*2705䡠pVIPR Tested on
B*2709䡠pVIPR Tested on
B*2705䡠pLMP2 Tested on
B*2709䡠pLMP2
MP 70 48
b
33 39 47
AS 38 68 63 38 27
AB 5 60 51 41 33
MP VPAC 7 42 45 64 41
MP VPAC 22 52 50 78 62
EP VIP 37 67 62 20 31
a
All donors are HLA-B*2705-positive AS patients.
b
Percentage of specific lysis of T2-B*2705 or T2-B*2709 cells after background subtraction.
TABLE V
pVIPR-stimulated CTL and assessment of pVIPR/pVIPR-pArg
3
cross-reactivity
CTL
a
Tested on
B*2705䡠pVIPR Tested on
B*2709䡠pVIPR Tested on
B*2705䡠pVIPR-pArg
3
Tested on
B*2709䡠pVIPR-pArg
3
EP 76 41
b
413 7
LV 72 73 57 3 6
PM 65 54 80 4 NT
c
PM 46 44 48 9 7
PM 45 47 63 13 NT
MP VPAC 7 57 51 23 11
PM 53 68 63 28 36
LV 1 58 55 28 NT
AB 4 42 52 49 NT
AB 5 60 51 59 NT
PM 63 75 62 10 77
a
All donors are HLA-B*2705-positive AS patients except LV who is an HLA-B*2702-positive AS patient.
b
Percentage of specific lysis of T2-B*2705 or T2-B*2709 cells after background subtraction.
c
NT, not tested.
Molecular Mimicry in HLA-B27 Subtypes2968
FIG.4.Comparison of the lytic potential of pLMP2/pVIPR-cross-reactive CTL. Dose-response curves (effector:target ratio, 15:1) show the
cytotoxic activity of four pLMP2-stimulated (a– d) or pVIPR-stimulated (e– h) CTL lines against T2-B*2705 (a,c,e, and g) or T2-B*2709 (b,d,f, and
h) cells pulsed with pLMP2 or pVIPR at different concentrations. iand jshow the dose-response curves of the cytotoxic activity (effector:target
ratio, 3:1) of a representative clone (clone 8) derived from the CTL line MP VPAC 7. The spontaneous release of
51
Cr-labeled cells was less than
15%. One of three separate experiments is shown. The sequence of the TCR
␣
-chain expressed by clone 8 (k,upper line,inbold) as well as that of
cross-reactive clones (k,lower line,initalics) with specificity for peptides derived from the BZLF1 protein of EBV and a serine/threonine kinase
(see text) (34) are also depicted.
Molecular Mimicry in HLA-B27 Subtypes 2969
the residues p1–p3 are very similar. The reactivities of pVIPR-
specific CTL with pVIPR- or pVIPR-pArg
3
-loaded B*2705 mol-
ecules corroborate these conclusions (Tables III, IV, and V).
They show that the majority of CTL is negatively influenced by
the pLys
3
Arg replacement, suggesting that the epitopes of their
TCR encompass the area around this residue. TCR
␣
-chain-
binding footprints are generally located above the N-terminal
half of the peptides and the surrounding binding groove resi-
dues (42–46). Therefore, an involvement of the N-terminal half
of the pLMP2 and pVIPR peptides as putative TCR epitopes
suggests that TCR
␣
-chains may contribute to the recognition
process.
This assumption is supported by our finding that CTL clones
derived from the MP VPAC 7 line use the same TCR
␣
-chain
CDR3 and J-region motifs as clones shown to recognize two
peptides derived from a viral (BZLF1 protein of EBV, RAK-
FKQLL) and a self-protein (serine/threonine kinase, RSKFR-
QIV) (34) (Fig. 4). These otherwise completely unrelated CTL
clones (HLA-B27- or HLA-B8-restricted, respectively) each rec-
ognize peptides with pArg
1
. It appears therefore plausible that
charge complementarity between pArg
1
and aspartic acid res-
idues in the TCR
␣
-chain CDR3 regions of both types of CTL
could be crucial to the recognition process, supporting our
assumption of the particular relevance of the N-terminal half of
the peptides in pLMP2/pVIPR functional and structural mim-
icry. However, it is currently unknown whether a relationship
exists between the observed cross-reactivity of the two HLA-
B27-presented peptides, the development of differential T cell
repertoires in the two subtypes, and AS pathogenesis. It even
remains possible that pLMP2 and pVIPR are not the peptides
that exhibit molecular mimicry relevant in the context of AS
but other, possibly heteroclitic, peptides that exhibit cross-
reactivity with the former. Nevertheless, these two peptides
allow elucidation of the structural basis of TCR cross-reactivity
in HLA-B27 subtypes that are differentially associated with an
autoimmune disease (1–4).
MHC class II molecules are more likely to generate examples
of structural mimicry of T cell epitopes than class I antigens (8,
9), because peptides are anchored not only at the termini but
also at several positions along an MHC class II binding groove.
This allows the formation of highly similar TCR epitopes even
when distantly related peptides are presented by different HLA
class II molecules (9). However, functional molecular mimicry
has already been shown for MHC class I antigens in mice and
humans, and EBV-specific memory T cell clones recognizing
cross-reactive self-peptides have been found in the periphery
(34) and even in joints (47). Our results complement functional
data (4, 5, 34, 47) by providing a structural basis for CTL
cross-reactivity also for class I molecules, with the pLMP2 and
pVIPR peptides presented by HLA-B27 antigens as paradigms.
These results suggest that cellular mechanisms underlying
disease association with HLA class I antigens may not be
fundamentally different from those observed with HLA class
II molecules.
The four criteria required for a clear-cut case of molecular
mimicry as a cause for autoimmunity (48) are only partially
fulfilled in the context of HLA-B27. There is evidence for T cells
directed against a self-antigen (pVIPR) nearly exclusively in
individuals with the AS-associated subtype (4), and a viral
mimic (pLMP2) of this self-antigen has been identified (Ref. 4
and this study). On the other hand, although the presence of
EBV-specific and self-cross-reactive CTL has been demon-
strated in the periphery and joints, for example in patients
with oligoarticular juvenile idiopathic arthritis (49), an epide-
miological association between AS and EBV has not been found
so far and is hard to demonstrate given the fact that about 90%
of humans get this infection during the first decades of life (35).
Furthermore, no animal models are available involving pres-
entation of pVIPR and pLMP2 in the context of HLA-B27. It is
currently unknown whether the structural mimicry described
here and the functional dichotomy between B*2705 and
B*2709 extend to HLA-B27 subtypes such as B*2704 and
B*2706, which are differentially associated with AS as well (1).
Our findings might be relevant not only in the context of
autoimmune diseases but are likely to influence also our un-
derstanding of conditions leading to acceptance or rejection of
transplants (50), in particular bone marrow grafts from unre-
lated donors (51). In addition, polymorphism of HC residue 116
has already been shown to exert an influence on the progres-
sion to AIDS among HIV-1
⫹
patients with different HLA-B35
subtypes; alleles with a Ser
116
(B*3501 and B*3508) were as-
sociated with slow progression, whereas B*3503 (Phe
116
but
otherwise identical to B*3501) was associated with more rapid
progression (52). This difference has been suggested to be due
to differential binding of HIV-derived peptides by these sub-
types (52).
Structural data comparing the B*3501/B*3503 pair are not
available, but a direct effect of residue 116 polymorphism on
the repertoire of bound peptides through interaction with the
C-terminal peptide side chain (3, 53, 54) or even with an amino
acid within the middle of a peptide such as pVIPR (5) or pLMP2
(this study) has been demonstrated. Furthermore, the peptide
repertoire may also be influenced indirectly by residue 116
polymorphism. This can be achieved through differential de-
pendence on the chaperone tapasin for loading of peptide cargo
(54–59). For example, B*4405 can be relatively efficiently
loaded with peptides also in the absence of tapasin, whereas
B*4402 (differing from B*4405 only in residue 116) exhibits a
complete dependence on this chaperone (54). The surface ex-
pression of tapasin-dependent HLA class I alleles may be dras-
tically impaired, with obvious consequences for the immune
response, when tapasin function is inhibited, as by the viral
US3 protein following an infection with cytomegalovirus (59).
Although as yet unproven, it remains a distinct possibility that
B*2705 and B*2709 also exhibit differential tapasin depend-
ence (60). In conclusion, residue 116-dependent differential
peptide presentation and the ensuing distinct CTL responses
could well serve to explain several of the HLA class I subtype-
dependent immune phenomena observed in AS as well as in the
other disease states mentioned above.
Acknowledgments—We thank all patients and healthy probands for
participation in this study and are grateful to Dr. U. Mu¨ ller for help
with x-ray data collection at BESSY II.
REFERENCES
1. Ramos, M., and Lo´ pez de Castro, J. A. (2002) Tissue Antigens 60, 191–205
2. D’Amato, M., Fiorillo, M. T., Carcassi, C., Mathieu, A., Zuccarelli, A., Bitti,
P. P., Tosi, R., and Sorrentino, R. (1995) Eur. J. Immunol. 25, 3199–3201
3. Ramos, M., Paradela, A., Vazquez, M., Marina, A., Vazquez, J., and Lo´ pez de
Castro, J. A. (2002) J. Biol. Chem. 277, 28749–28756
4. Fiorillo, M. T., Maragno, M., Butler, R., Dupuis, M. L., and Sorrentino, R.
(2000) J. Clin. Invest. 106, 47–53
5. Hu¨ lsmeyer, M., Fiorillo, M. T., Bettosini, F., Sorrentino, R., Saenger, W.,
Ziegler, A., and Uchanska-Ziegler, B. (2004) J. Exp. Med. 199, 271–281
6. Oldstone, M. B. (1987) Cell 50, 819– 820
7. Benjamin, R., and Parham, P. (1990) Immunol. Today 11, 137–142
8. Wucherpfennig, K. W. (2001) J. Autoimmun. 16, 293–302
9. Lang, H. L., Jacobsen, H., Ikemizu, S., Andersson, C., Harlos, K., Madsen, L.,
Hjorth, P., Sondergaard, L., Svejgaard, A., Wucherpfennig, K., Stuart, D. I.,
Bell, J. I., Jones, E. Y., and Fugger, L. (2002) Nat. Immunol. 3, 940–943
10. Geczy, A. F., and Yap, J. (1982) J. Rheumatol. 9, 97–100
11. Schwimmbeck, P. L., Yu, D. T., and Oldstone, M. B. (1987) J. Exp. Med. 166,
173–181
12. Ramos, M., Alvarez, I., Sesma, L., Logean, A., Rognan, D., and Lo´pez de
Castro, J. A. (2002) J. Biol. Chem. 277, 37573–37581
13. Tiwana, H., Walmsley, R. S., Wilson, C., Yiannakou, J. Y., Ciclitira, P. J.,
Wakefield, A. J., and Ebringer, A. (1998) Br. J. Rheumatol. 37, 525–531
14. Stone, M. A., Payne, U., Schentag, C., Rahman, P., Pacheco-Tena, C., and
Inman, R. D. (2004) Rheumatology 43, 148–155
15. Wucherpfennig, K. W., and Strominger, J. L. (1995) Cell 80, 695–705
Molecular Mimicry in HLA-B27 Subtypes2970
16. Lombardi, G., Germain, C., Uren, J., Fiorillo, M. T., du Bois, R. M., Jones-
Williams, W., Saltini, C., Sorrentino, R., and Lechler, R. (2001) J. Immunol.
166, 3549–3555
17. Kalams, S. A., Johnson, R. P., Trocha, A. K., Dynan, M. J., Ngo, H. S., D’Aquila,
R. T., Kurnick, J. T., and Walker, B. D. (1994) J. Exp. Med. 179, 1261–1271
18. Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276, 307–326
19. Collaborative Computational Project Number 4 (1994) Acta Crystallogr. Sect.
D Biol. Crystallogr. 50, 760–763
20. Bru¨ nger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-
Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read,
R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr.
Sect. D Biol. Crystallogr. 54, 905–921
21. Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S., and Dodson, E. J.
(1999) Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 247–255
22. Jones, T. A., and Kjeldgaard, M. (1997) Methods Enzymol. 277, 173–208
23. Perrakis, A., Morris, R., and Lamzin, V.S. (1999) Nat. Struct. Biol. 6, 458– 463
24. Winn, M. D., Isupov, M. N., and Murshudov, G. N. (2001) Acta Crystallogr.
Sect. D Biol. Crystallogr. 57, 122–133
25. Merritt, E. A. (1999) Acta Crystallogr. Sect. D Biol. Crystallogr. 55, 1109–1117
26. Hooft, R. W., Vriend, G., Sander, C., and Abola, E. E. (1996) Nature 381, 272
27. Nicholls, A., Sharp, K. A., and Honig, B. (1991) Proteins 11, 281–296
28. Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946–950
29. Sanner, M. F., Olson, A. J., and Spehner, J. C. (1996) Biopolymers 38, 305–320
30. Merritt, E. A., and Bacon, D. J. (1997) Methods Enzymol. 277, 505–527
31. Madden, D. R. (1995) Annu. Rev. Immunol. 13, 587–622
32. Brooks, J. M., Murray, R. J., Thomas, W. A., Kurilla, M. G., and Rickinson,
A. B. (1993) J. Exp. Med. 178, 879–887
33. Contu, L., Arras, M., Carcassi, C., La Nasa, G., and Mulargia, M. (1992) Tissue
Antigens 40, 165–174
34. Misko, I. S., Cross, S. M., Khanna, R., Elliott, S. L., Schmidt, C., Pye, S. J., and
Silins, S. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96, 2279–2284
35. Moss, D. J., Burrows, S. R., Silins, S. L., Misko, I., and Khanna, R. (2001)
Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 475–488
36. Earnest, T., Fauman, E., Craik, C. S., and Stroud, R. (1991) Proteins 10,
171–187
37. Tilton, R. F., Dewan, J. C., and Petsko, G. A. (1992) Biochemistry 31,
2469–2481
38. Wagner, G., Hyberts, S. G., and Havel, T. F. (1992) Annu. Rev. Biophys.
Biomol. Struct. 21, 167–198
39. Stollar, E. J., Mayor, U., Lovell, S. C., Federici, L., Freund, S. M. V., Fersht,
A. R., and Luisi, B. F. (2003) J. Biol. Chem. 278, 43699–43708
40. Liu, Y., Zhang, X., Yoshida, T., and La Mar, G. N. (2004) Biochemistry 43,
10112–10126
41. Po¨ hlmann, T., Bo¨ckmann, R. A., Grubmu¨ ller, H., Uchanska-Ziegler, B.,
Ziegler, A, and Alexiev U. (2004) J. Biol. Chem. 279, 28197–28201
42. Garboczi, D. N., Ghosh, P., Utz, U., Fan, Q. R., Biddison, W. E., and Wiley,
D. C. (1996) Nature 384, 134–141
43. Garcia, K. C., Degano, M., Stanfield, R. L., Brunmark, A., Jackson, M. R.,
Peterson, P. A., Teyton, L., and Wilson, I. A. (1996) Science 274, 209–219
44. Reiser, J. B., Darnault, C., Guimezanes, A., Gregoire, C., Mosser, T., Schmitt-
Verhulst, A. M., Fontecilla-Camps, J. C., Malissen, B., Housset, D., and
Mazza, G. (2000) Nat. Immunol. 1, 291–297
45. Housset, D., and Malissen, B. (2003) Trends Immunol. 24, 429– 437
46. Kjer-Nielsen, L., Clements, C. S., Purcell, A. W., Brooks, A. G., Whisstock,
J. C., Burrows, S. R., McCluskey, J., and Rossjohn, J. (2003) Immunity 18,
53–64
47. Selin, L. K., and Welsh, R. M. (2004) Immunity 20, 5–16
48. Rose, N. R., and Bona, C. (1993) Immunol. Today 14, 426– 430
49. Massa, M., Mazzoli, F., Pignatti, P., De Benedetti, F., Passalia, M., Viola, S.,
Samodal, R., La Cava, A., Giannoni, F., Ollier, W., Martini, A., and Albani,
S. (2002) Arthritis Rheum. 46, 2721–2729
50. Doxiadis, I. I., Smits, J. M., Schreuder, G. M., Persijn, G. G., van Houwelingen,
H. C., van Rood, J. J., and Claas, F. H. (1996) Lancet 348, 850–853
51. Ferrara, G. B., Bacigalupo, A., Lamparelli, T., Lanino, E., Delfino, L., Mora-
bito, A., Parodi, A. M., Pera, C., Pozzi, S., Sormani, M. P., Bruzzi, P., Bordo,
D., Bolognesi, M., Bandini, G., Bontadini, A., Barbanti, M., and Frumento,
G. (2001) Blood 98, 3150–3155
52. Gao, X., Nelson, G. W., Karacki, P., Martin, M. P., Phair, J., Kaslow, R.,
Goedert, J. J., Buchbinder, S., Hoots, K., Vlahov, D., O’Brien, S. J., and
Carrington, M. (2001) N. Engl. J. Med. 344, 1668–1675
53. Hu¨ lsmeyer, M., Hillig, R. C., Volz, A., Ru¨ hl, M., Schro¨ der, W., Saenger, W.,
Ziegler, A., and Uchanska-Ziegler, B. (2002) J. Biol. Chem. 277,
47844–47853
54. Zernich, D., Purcell, A. W., Macdonald, W. A., Kjer-Nielsen, L., Ely, L. K.,
Laham, N., Crockford, T., Mifsud, N. A., Bharadwaj, M., Chang, L., Tait,
B. D., Holdsworth, R., Brooks, A. G., Bottomley, S. P., Beddoe, T., Peh, C. A.,
Rossjohn, J., and McCluskey, J. (2004) J. Exp. Med. 200, 13–24
55. Peh, C. A., Burrows, S. R., Barnden, M., Khanna, R., Cresswell, P., Moss, D. J.,
and McCluskey, J. (1998) Immunity 8, 531–542
56. Turnquist, H. R., Thomas, H. J., Prilliman, K. R., Lutz, C. T., Hildebrand,
W. H., and Solheim, J. C. (2000) Eur. J. Immunol. 30, 3021–3028
57. Turnquist, H., Schenk, E. L., McIlhaney, M. M., Hickman, H. D., Hildebrand,
W. H., and Solheim, J. C. (2002) Immunogenetics 53, 830–834
58. Hildebrand, W. H., Turnquist, H. R., Prilliman, K. R., Hickman, H. D., Schenk,
E. L., McIlhaney, M. M., and Solheim, J. C. (2002) Hum. Immunol. 63,
248–255
59. Park, B., Kim, Y., Shin, J., Lee, S., Cho, K., Fruh, K., Lee, S., and Ahn, K.
(2004) Immunity 20, 71–85
60. Uchanska-Ziegler, B., Alexiev, U., Hillig, R., Hu¨ lsmeyer, M., Po¨ hlmann, T.,
Saenger, W., Volz, A., and Ziegler, A. (2004) in Proceedings of the 13
th
International Histocompatibility Workshop and Congress (Hansen, J.A.,
and Dupont, B., eds) International Histocompatibility Working Group
Press, Seattle, in press
Molecular Mimicry in HLA-B27 Subtypes 2971