





Available via license: CC BY-NC-ND 4.0
Content may be subject to copyright.
Study of novel triazolo-benzodiazepine analogues as antidepressants
targeting by molecular docking and ADMET properties prediction
Assia Belhassan
a
,
b
, Hanane Zaki
b
,
c
, Mohamed Benlyas
c
, Tahar Lakhlifi
a
,
Mohammed Bouachrine
a
,
b
,
*
a
MCNS Laboratory, Faculty of Science, Moulay Ismail University, Meknes, Morocco
b
Materials, Environment &Modeling Laboratory, High School of Technology, Moulay Ismail University, Meknes, Morocco
c
Biology Environment and Health Laboratory, Faculty of Science and Technics, Moulay Ismail University, Errachdia, Morocco
ARTICLE INFO
Keywords:
Theoretical chemistry
Pharmaceutical chemistry
Bioinformatics
Biochemistry
Triazolo-benzodiazepine
Antidepressant activity
Molecular docking
ADMET properties
Nortriptyline
ABSTRACT
In this study, we have selected a series of a new family of molecules bearing Triazolo-benzodiazepines, an eleven
membered heterocyclic ring has been studied for antidepression activity. Docking studies suggested that all the
eleven ligands interacted well within active site of Drosophila melanogaster dopamine transporter (dDAT) (PDB
ID: 4M48). Most ligands formed H-bond with amino acid Phe43, Asp46, Asp475, Tyr123, Ser421 and/or Gln316
and also exhibited Pi and Pi-Pi interactions with amino acid residues Tyr124, Phe319, Phe43, Phe325, Ala479 and
Val120. In silico ADME evaluations of compounds showed more than 96% intestinal absorption for all com-
pounds. During in vitro Toxicity properties prediction, the Triazolo-benzodiazepines derivatives: M
1
,M
2
,M
3
and
M
11
showed less toxicity than the other studied molecules against algae, for daphnia the molecules M
1
,M
2
,M
3
,
M
8
,M
10
and M
11
showed less toxicity than the reference molecule (Nortriptyline).
1. Introduction
Diazepines are a well-known class of heterocycles and they have
gained importance since 1957, when the chlordiazepoxide (first benzo-
diazepine) was synthesized and studied in terms of psychotropic activity
[1,2,3]. Actually, they possess a wide spectrum of biological activity
including anxiolytic, hypnotic, sedative, anticonvulsant, skeletal,
amnestic and muscle relaxant properties [4,5,6,7].
Triazolo-benzodiazepines analogues are a key structural motif in
numerous therapeutics that have sedative, muscle relaxant, and anti-
tumor activities [8,9]. Alprazolam, adinazolam and estazolam are
commercially available chemical drugs based on triazolo-benzodiazepine
scaffold that widely used as anxiolytic and sedative agents [10,11,12,
13]. Some triazolo-benzodiazepine derivatives have been reported to be
weakly bound to the benzodiazepine receptor and prevent serine prote-
ase [14,15].
So, due to the therapeutic and biological applications of this class of
compounds, the study of type of interactions between these molecules
and protein targetingby molecular docking methods for the prediction of
the activity is definitely of great importance.
Molecular docking turns out to be a reliable method for preliminary
evaluation of binding affinity and prediction of intermolecular in-
teractions of novel compounds with receptors [16]. Nowadays, this
method has become indispensable for studying protein-ligand in-
teractions. Docking method can produce significant knowledge for
complex systems, which complements experimentally achievable data.
Molecular docking simulations have found widespread application for
virtual screening and pose prediction of new or non-synthesized com-
pounds [17,18,19,20].
Molecular docking studies were focused on the dopamine trans-porter
(DAT). This transmembrane protein is responsible for reup-take of
dopamine from the synaptic cleft. DAT inhibitors are used in the treat-
ment of depression due to the increased level of dopamine in the synaptic
cleft as well as in adjuvant therapy of Parkinson's Disease (PD) [21].
In this paper, a new family of Triazolo-benzodiazepines (Fig. 1) was
docked to neurotransmitter trans-porter (DAT). We predict and interpret
thebinding affinity and intermolecular interactions of complexes formed
by docking of these molecules on DAT protein, so as to gain insight if
those newly synthesized compounds could be of use as therapeutics in
medicine.
* Corresponding author.
E-mail address: m.bouachrine@est-umi.ac.ma (M. Bouachrine).
Contents lists available at ScienceDirect
Heliyon
journal homepage: www.heliyon.com
https://doi.org/10.1016/j.heliyon.2019.e02446
Received 9 July 2019; Received in revised form 24 August 2019; Accepted 4 September 2019
2405-8440/©2019 The Author(s). Published by Elsevier Ltd. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-
nc-nd/4.0/).
Heliyon 5 (2019) e02446
2. Material and methods
2.1. Data collection
2.1.1. Ligands
In the present study, a series of 11 selected Triazolo-benzodiazepines
derivatives were taken from literature (Table 1)[22], these molecules
were considered to molecular docking study. The selected
Triazolo-benzodiazepines were prepared by Asgari et al. from simple
methods and substrates [22].
2.1.2. Receptor: dopamine transporter DAT
The dopamine trans-porter (DAT) is transmembrane protein.
Drosophila melanogaster dopamine transporter (dDAT) is a proven
model of DAT, which is 50% similar to mammalian DAT sequence, and is
used in research into the mechanism of action of many compounds. Thus,
in our docking studies we made use of the X-ray structure of dDAT in
complex with the tricycilcanitide-pressant nortriptyline, which indicate
primary binding site (PDB code: 4M48) [16].
2.2. Molecular docking studies
Molecular docking turns out to be a reliable method for pre-liminary
evaluation of binding affinity and prediction of intermolecular in-
teractions of novel compounds with receptors. We decided to use
neurotrans-mitter transporters (DAT) for docking study. In general, our
research is based on crystal structures of receptors with bound ligand
molecules. This structure has been obtained from X-ray crystal data of
RCSB Protein Data Bank (PDB). In the majority of selected structures, co-
crystallized ligand molecules are known drugs with proven action, and
thus determine the binding site location in DAT as well as serve as ref-
erences in our considerations.
In this research space, a docking process is launched for each studied
molecule; the bioactive conformations were simulated using Autodock
vina and Autodock tools 1.5.6 [23]. The results were analyzed using
Discovery studio 2016 [24] and PyMol [25] softwares.
2.3. ADME and toxicity prediction
Absorption, distribution, metabolism, excretion and toxicity are pre-
dicted for the 11 selected Triazolo-benzodiazepines derivatives using Pre
ADMET predictor server [26,27].
3. Results and discussion
3.1. Molecular docking
The top-scoring pose of each molecule is selected according to the
best interaction energy with the Drosophila melanogaster dopamine
transporter (dDAT) (Table 2).
The best energies of interaction with the Drosophila melanogaster
dopamine transporter (dDAT) (lowest energy level) are observed for the
ligand M
10
, whereas the ligand M
6
is the least stable ligand in the list of
studied molecules (Table 2). We can also observe that all complex formed
by studied compounds and dDAT are more stable than the complex
formed with the reference molecule (Nortriptyline) except for two
studied molecules: M
2
and M
6
.
The result of the re-docked Nortriptyline molecule and its position in
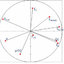
the PDB structure of protein dDAT is shown in Fig. 2.
Nortriptyline is involved in Pi-Pi T-shaped interactions with Tyr124
as well as alkyl and Pi-alkyl interactions with Ala479, and Pi-sigma in-
teractions with Val120. The N-methylpropanamine chain of nortriptyline
enables formation of carbon hydrogen bonds with Asp46, Ser421 and
Phe319, while conventional hydrogen bon and Pi-donor hydrogen bond
are created only with Phe43 because nitrogen atom (N) is present in this
position. The Van der Waals interactions are observed with Ile 483, Tyr
Fig. 1. Chemical structures of studied compounds.
Table 1
Chemical structures of the 11 selected Triazolo-benzodiazepines derivatives.
NRR
1
R
2
R
3
1 Cyclohexyl O–CH
3
HH
2 Cyclohexyl O–CH
3
O–CH
3
H
3 Cyclohexyl Cl H H
4t-Butyl CH
3
HH
5t-Butyl H H CH
3
6t-Butyl O–CH
3
HH
7t-Butyl O–CH
3
O–CH
3
H
8t-Butyl Cl H H
9t-Butyl H H F
10 Cyclohexyl F H H
11 1,1,3,3-tetramethyl-butyl F H H
Table 2
Comparison of Autodock score (kcal/mol) of the 9 best poses obtained by docking of 11 selected Triazolo-benzodiazepines derivatives and the re-docking of reference
molecule (Nortriptyline) with dDAT.
Ligands 123456789
Ref: Nortriptyline -10.0 -9.3 -9.2 -9.0 -8.8 -8.7 -8.7 -8.5 -8.0
M
1
-10.0 -9.8 -9.2 -9.1 -9.0 -8.9 -8.8 -8.7 -8.7
M
2
-9.9 -9.5 -9.4 -9.2 -9.1 -8.9 -8.9 -8.8 -8.7
M
3
-10.6 -10.0 -10.0 -9.9 -9.8 -9.6 -9.5 -9.4 -9.4
M
4
-10.5 -9.4 -9.3 -9.3 -9.3 -9.2 -9.2 -9.1 -8.8
M
5
-10.4 -9.3 -9.3 -9.3 -9.1 -8.7 -8.7 -8.7 -8.6
M
6
-9.6 -9.2 -9.1 -9.1 -9.0 -9.0 -8.9 -8.9 -8.7
M
7
-10.0 -9.9 -9.5 -9.5 -9.4 -9.2 -9.0 -9.0 -8.9
M
8
-10.4 -9.8 -9.5 -9.4 -9.4 -9.3 -9.3 -9.2 -8.9
M
9
-10.3 -9.4 -9.4 -9.3 -9.3 -9.1 -9.0 -8.9 -8.8
M
10
-12.3 -10.6 -10.5 -10.3 -10.0 -10.0 -9.8 -9.6 -9.5
M
11
-10.1 -10.1 -9.9 -9.5 -9.3 -9.1 -8.8 -8.8 -8.7
A. Belhassan et al. Heliyon 5 (2019) e02446
2
123 and Gly 425 amino acids.
The docking result of 11 selected Triazolo-benzodiazepines de-
rivatives and dDAT is shown in Fig. 3. And the comparison of these re-
sults and the result of the re-docked Nortriptyline molecule and its
position in the PDB structure of protein dDAT is shown in Table 3.
Visual inspection of the docked poses of molecule M
11
clearly in-
dicates similarity between binding modes and interactions of this mole-
cule and the reference molecule (Nortriptyline) with dDAT. Both of them
form carbon hydrogen bonds with Asp46, while conventional hydrogen
hydrogen bond is formed with Phe43. Moreover, Tyr124 is bonded with
M
3
,M
4
,M
5
,M
7
,M
8
,M
9
and M
9
by Pi-Pi interactions, which play a similar
role in the binding of docked Nortriptyline molecule. All orientations of
the discussed Triazolo-benzodiazepines derivatives are stabilized in the
dDAT cavity by weak hydrophobic interactions with Val120 and Ala479
in a similar manner to docked Nortriptyline except the two molecules M
5
and M
9
.
The similarities between interactions of 11 Triazolo-benzodiazepines
derivatives and reference molecule are retained to usas therapeutics in
medicine to treat the depression.
3.2. ADME, toxicity and drug likeness prediction
Absorption, distribution, metabolism, excretion, toxicity and drug
likeness are predicted for the 11selected Triazolo-benzodiazepines de-
rivatives using Pre ADMET predictor server, and the results are presented
in Tables 4and 5.
The analysis of predicted ADME properties results (Table 4) shows
that: the eleven molecules have different predicted in vivo blood-brain
barrier penetration, the molecules M
1
,M
2
,M
3
and M
10
have highest
penetration (0.318, 0.320, 0.340 and 0.331, respectively) in comparison
with the other molecules, whereas the molecule M
4
has a very low
permeability (0.122). All these values are largely insufficient; in fact
blood-brain barrier penetration of antidepressant molecules can reach for
example in Nortriptyline 13.406.
All the molecules can't inhibit or be substrate for cytochromes
CYP_2C19, CYP_2C9 and CYP_2D6 while they inhibit and substrate cy-
tochrome CYP_3A4. These molecules have a high absorption which can
exceed 96% for all the molecules, which is important for oral adminis-
tration. A percent of plasma proteinbinding more than 80% is noted for
all molecules which mean that 20% of the fraction of these molecules can
actually give the pharmacological effect. This doesn't prevent that pro-
tein binding can influence the drug's biological half-life. The bound
portion may act as a reservoir or depot from which the drug is slowly
released as the unbound form.
The results of the prediction of the toxicity presented in Table 5 show
that these molecules show a very low toxicity on the algae and daphnia,
and a negative toxicity according to the four Ames tests (in vitro Ames
test in TA100 strain (Metabolic activation by rat liver homogenate), in
vitro Ames test in TA100 strain (No metabolic activation), in vitro Ames
test in TA1535 strain (Metabolic activation by rat liver homogenate), in
vitro Ames test in TA1535 strain (No metabolic activation)) except M
4
,
M
5
,M
6
,M
7
,M
8
and M
9
whom has a positive toxicity on in vitro Ames test
in TA100 strain (Metabolic activation by rat liver homogenate).
The Ames's mutagenicity test that uses several strains of the bacte-
rium Salmonella typhimurium that carry mutations in genes involved in
histidine synthesis, so that they require histidine for growth show that
the molecule M
2
can induce mutations, and none of these molecules
present a risk of carcinogenicity neither in the rat nor in the mouse, and
Fig. 2. Types of interactions between the dDAT (PDB code: 4M48) and Nortriptyline.
A. Belhassan et al. Heliyon 5 (2019) e02446
3
Fig. 3. Types of interactions between the dDAT (PDB code: 4M48) and the 11 selected Triazolo-benzodiazepines derivatives.
A. Belhassan et al. Heliyon 5 (2019) e02446
4
Table 3
Comparison of interactions formed by docking of 11 selected Triazolo-benzodiazepines derivatives and the re-docking of reference molecule (Nortriptyline) with dDAT.
Type of interactions Residues Molecules
Ref: Nortriptyline M
1
M
2
M
3
M
4
M
5
M
6
M
7
M
8
M
9
M
10
M
11
Hydrogen
Bonds
Conventional
H-Bond
PHE43 X X
GLN316 X
ASP46 X X X
TYR123 X X X
SER421 X
ASP475 X
Carbon -H-Bond SER421 X X X X
ASP46 X X X
PHE319 X X
ALA479 X
SER320 X
ASP475 X
TYR123 X
ALA44 X
SER124 X
Pi-Donor-H-Bond PHE43 X
Hydrophobic
interactions
Pi-Pi TYR124 X X X X X X X X
PHE319 X X X X X
PHE43 X X
PHE325 X
Alkyl VAL120 X X X X X
ALA479 X
ILE483 X
ILE127 X
ARG52 X X
ALA44 X X
ALA48 X
TYR124 X
Pi-Alkyl ALA479 X X X X
PHE319 X X X
PHE325 X
TRP51 X X
TYR123 X
TYR124 X X X
PHE471 X
VAL120 X
Pi-Sigma VAL120 X X X X X X X
PHE43 XX
Table 4
Predicted ADME properties of the 11 studied compounds in comparison with the reference drug.
Ref M
1
M
2
M
3
M
4
M
5
M
6
M
7
M
8
M
9
M
10
M
11
BBB 13.406 0.318 0.320 0.340 0.122 0.159 0.245 0.197 0.277 0.180 0.331 0.246
BS 66.488 93.970 52.011 59.350 247.204 77.602 326.688 181.526 206.459 368.859 189.694 157.659
CYP_2C19_inhibition Inhibitor Non Non Non Non Non Non Non Non Non Non Non
CYP_2C9_inhibition Non Non Non Non Non Non Non Non Non Non Non Non
CYP_2D6_inhibition Inhibitor Non Non Non Non Non Non Non Non Non Non Non
CYP_2D6_substrate Substrate Non Non Non Non Non Non Non Non Non Non Non
CYP_3A4_inhibition Non Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor Inhibitor
CYP_3A4_substrate Non Weakly Substrate Weakly Substrate Substrate Substrate Substrate Substrate Substrate Weakly Substrate
HIA 100 96.828 97.356 96.548 96.466 96.466 97.004 97.525 96.457 96.506 96.438 96.440
MDCK 96.879 1.121 0.143 3.675 24.933 112.593 6.772 1.476 9.663 27.802 2.223 0.043
Pgp_I Inhibitor Non Inhibitor Inhibitor Non Non Inhibitor Inhibitor Inhibitor Non Non Inhibitor
PPB 100 90.524 89.190 91.919 88.306 87.741 83.385 80.719 87.854 88.330 89.897 89.716
PWS 2.941 2.472 1.721 0.520 9.662 18.801 22.013 15.382 4.624 21.762 1.755 0.514
SKlogD_value 3.500 3.554 3.5489 4.269 3.407 3.3850 2.876 2.872 3.592 3.046 3.735 4.600
SKlogP_value 4.844 3.554 3.5489 4.269 3.407 3.3850 2.876 2.872 3.592 3.046 3.735 4.600
SKlogS_buffer -3.621 -3.676 -3.961 -3.880 -3.213 -3.716 -3.109 -3.394 -3.312 -3.043 -3.359 -3.468
SKlogS_pure -4.975 -5.256 -5.441 -5.938 -4.621 -4.332 -4.280 -4.466 -4.962 -4.272 -5.393 -5.955
BBB ¼in vivo blood-brain barrier penetration (C.brain/C.blood), BS ¼Buffer Solubility (mg/l), CYP2C19 ¼cytochrome P4502C19, CYP2C9 ¼cytochrome P4502C9,
CYP3A4 ¼cytochrome P4503A4, CYP2D6 ¼cytochrome CYP2D6, PgP I ¼P-glycoprotein inhibition, PPB ¼Plasma Protein Binding %, PWS ¼Pure Water Solubility
(mg/l), HIA ¼Human intestinal absorption %, MDCK ¼in vitro MDCKcellpermeability (Mandin Darby Canine Kidney (nm/sec)).
A. Belhassan et al. Heliyon 5 (2019) e02446
5
all present a medium risk to inhibit HERG (Human ether-a-go-go related
gene channel). It should be noted that all these values are predicted.
4. Conclusion
In this study, the docking study was performed to elucidate the type of
interactions between 11 selected Triazolo-benzodiazepines derivatives
and Drosophila melanogaster dopamine transporter (dDAT), the results
show that all the eleven ligands interacted well within active site of dDAT
(PDB ID: 4M48); the molecules showed promising in silico results as
indicated by their high protein–ligand interaction energy. The studied
compounds are screened by ADME and Toxicity properties; these mole-
cules are predicted to have more than 96% intestinal absorption for all
compounds. During in vitro Toxicity properties prediction, the Triazolo-
benzodiazepines derivatives: M
1
,M
2
,M
3
,M
8
,M
10
and M
11
showed less
toxicity than the reference molecule (Nortriptyline) against daphnia.
A deep investigation of in vitro activity supported by docking results
and in silico ADMET results clearly suggested that these molecules could
be of use as therapeutics in medicine to treat the depression.
Declarations
Author contribution statement
Mohammed Bouachrine, Assia Belhassan, Hanane Zaki, Mohamed
Benlyas, Tahar Lakhlifi: Conceived and designed the experiments; Per-
formed the experiments; Analyzed and interpreted the data; Contributed
reagents, materials, analysis tools or data; Wrote the paper.
Funding statement
This research did not receive any specific grant from funding agencies
in the public, commercial, or not-for-profit sectors.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
We are grateful to the “Association Marocaine des Chimis-
tesTh
eoriciens”(AMCT) for its pertinent help concerning the programs.
References
[1] T.A. Ban, The role of serendipity in drug discovery, Dialogues Clin. Neurosci. 8
(2006) 335–344. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3181823/.
(Accessed 3 May 2019).
[2] L.H. Sternbach, E. Reeder, Quinazolines and 1, 4-benzodiazepines. IV. 1, 2
transformations of 7-Chloro-2-methylamino-5-phenyl-3H-1, 4-benzodiazepine 4-
oxide3, J. Org. Chem. 26 (1961) 4936–4941.
[3] G. Zbinden, L.O. Randall, Pharmacology of benzodiazepines: laboratory and clinical
correlations. Adv. Pharmacol., Elsevier, 1967, pp. 213–291.
[4] H. Mofakham, A. Shaabani, S. Mousavifaraz, F. Hajishaabanha, S. Shaabani,
S.W. Ng, A novel one-pot pseudo-five-component condensation reaction towards
bifunctional diazepine-tetrazole containing compounds: synthesis of 1H-tetrazolyl-
1H-1,4-diazepine-2,3-dicarbonitriles and 1H-tetrazolyl-benzo[b][1,4]diazepines,
Mol. Divers. 16 (2012) 351–356.
[5] N. Eleftheriadis, C.G. Neochoritis, C.A. Tsoleridis, J. Stephanidou-Stephanatou,
Z. Iakovidou-Kritsi, One-pot microwave assisted synthesis of new 2-alkoxycarbo-
nylmethylene-4-oxo-1,5-benzo-, naphtho-, and pyridodiazepines and assessment of
their cytogenetic activity, Eur. J. Med. Chem. 67 (2013) 302–309.
[6] S.-G. Huang, H.-F. Mao, S.-F. Zhou, J.-P. Zou, W. Zhang, Recyclable gallium(III)
triflate-catalyzed [4þ3] cycloaddition for synthesis of 2,4-disubstituted-3H-benzo
[b][1,4]diazepines, Tetrahedron Lett. 54 (2013) 6178–6180.
[7] B. Gerratana, Biosynthesis, synthesis, and biological activities of
pyrrolobenzodiazepines, Med. Res. Rev. 32 (2012) 254–293.
Table 5
Toxicity predicted of the 11 studied compounds in comparison with the reference drug.
ID Ref M
1
M
2
M
3
M
4
M
5
M
6
M
7
M
8
M
9
M
10
M
11
algae_at 0.008 0.019 0.013 0.011 0.053 0.064 0.055 0.040 0.037 0.076 0.025 0.011
Ames_test Mutagen Mutagen Non-mutagen Mutagen Mutagen Mutagen Mutagen Mutagen Mutagen Mutagen Mutagen Mutagen
Carcino_Mouse Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative
Carcino_Rat Positive Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative
daphnia_at 0.030 0.024 0.022 0.009 0.0434 0.044 0.078 0.070 0.029 0.059 0.020 0.012
hERG_inhibition Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk Medium_risk
TA100_10RLI Negative Negative Negative Negative Positive Positive Positive Positive Positive Positive Negative Negative
TA100_NA Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative
TA1535_10RLI Positive Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative
TA1535_NA Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative Negative
A. Belhassan et al. Heliyon 5 (2019) e02446
6
[8] Midazolam and Other Benzodiazepines jSpringerLink, (n.d.). https://link.spr
inger.com/chapter/10.1007/978-3-540-74806-9_16 (accessed April 28, 2019).
[9] L.H. Hurley, T. Reck, D.E. Thurston, D.R. Langley, K.G. Holden, R.P. Hertzberg,
J.R. Hoover, G. Gallagher Jr., L.F. Faucette, Pyrrolo [1, 4] benzodiazepine
antitumor antibiotics: relationship of DNA alkylation and sequence specificity to the
biological activity of natural and synthetic compounds, Chem. Res. Toxicol. 1
(1988) 258–268.
[10] S.M. Hanson, C. Czajkowski, Structural mechanisms underlying benzodiazepine
modulation of the GABAA receptor, J. Neurosci. 28 (2008) 3490–3499.
[11] P.J. Snyder, J. Werth, B. Giordani, A.F. Caveney, D. Feltner, P. Maruff, A method for
determining the magnitude of change across different cognitive functions in clinical
trials: the effects of acute administration of two different doses alprazolam, Hum.
Psychopharmacol. Clin. Exp. 20 (2005) 263–273.
[12] M. Watanabe, K. Maemura, K. Kanbara, T. Tamayama, H. Hayasaki, GABA and
GABA receptors in the central nervous system and other organs, in: K.W. Jeon (Ed.),
Int. Rev. Cytol., Academic Press, 2002, pp. 1–47.
[13] J. Levine, D.P. Cole, K.N.R. Chengappa, Anxiety disorders and major depression,
together or apart, Depress. Anxiety 14 (2001) 94–104.
[14] L. Bertelli, G. Biagi, I. Giorgi, O. Livi, C. Manera, V. Scartoni, C. Martini,
G. Giannaccini, L. Trincavelli, P.L. Barili, 1,2,3-Triazolo[1,5-a][1,4]- and 1,2,3-tri-
azolo[1,5-a][1,5]benzodiazepine derivatives: synthesis and benzodiazepine
receptor binding, Il Farmaco 53 (1998) 305–311.
[15] D.K. Mohapatra, P.K. Maity, M. Shabab, M.I. Khan, Click chemistry based rapid one-
pot synthesis and evaluation for protease inhibition of new tetracyclic triazole fused
benzodiazepine derivatives, Bioorg. Med. Chem. Lett 19 (2009) 5241–5245.
[16] A. Adamski, D. Kruszka, Z. Dutkiewicz, M. Kubicki, A. Gorczy
nski, V. Patroniak,
Novel family of fused tricyclic [1, 4] diazepines: design, synthesis, crystal structures
and molecular docking studies, Tetrahedron 73 (2017) 3377–3386.
[17] N. Mobaraki, B. Hemmateenejad, T.R. Weikl, A. Sakhteman, On the relationship
between docking scores and protein conformational changes in HIV-1 protease,
J. Mol. Graph. Model. 91 (2019) 186–193.
[18] D.B. Kitchen, H. Decornez, J.R. Furr, J. Bajorath, Docking and scoring in virtual
screening for drug discovery: methods and applications, Nat. Rev. Drug Discov. 3
(2004) 935.
[19] Z. Gaieb, S. Liu, S. Gathiaka, M. Chiu, H. Yang, C. Shao, V.A. Feher, W.P. Walters,
B. Kuhn, M.G. Rudolph, D3R Grand Challenge 2: blind prediction of protein–ligand
poses, affinity rankings, and relative binding free energies, J. Comput. Aided Mol.
Des. 32 (2018) 1–20.
[20] S. Gathiaka, S. Liu, M. Chiu, H. Yang, J.A. Stuckey, Y.N. Kang, J. Delproposto,
G. Kubish, J.B. Dunbar, H.A. Carlson, D3R grand challenge 2015: evaluation of
protein–ligand pose and affinity predictions, J. Comput. Aided Mol. Des. 30 (2016)
651–668.
[21] P. Huot, S.H. Fox, J.M. Brotchie, Dopamine reuptake inhibitors in Parkinson’s
disease: a review of nonhuman primate studies and clinical trials, J. Pharmacol.
Exp. Ther. 357 (2016) 562–569.
[22] M.S. Asgari, M. Soheilizad, P.R. Ranjbar, B. Larijani, R. Rahimi, M. Mahdavi, Novel
and efficient synthesis of triazolobenzodiazepine analogues through the sequential
Ugi 4CR-click-N-arylation reactions, Tetrahedron Lett. 60 (2019) 583–585.
[23] O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of docking
with a new scoring function, efficient optimization, and multithreading, J. Comput.
Chem. 31 (2010) 455–461.
[24] Dassault Syst
emes BIOVIA Discovery Studio Modeling Environment, Release 2017
Dassault Syst
emes, 2016. http://accelrys.com/products/collaborative-science/bio
via-discovery-studio/.
[25] W.L. DeLano, The PyMOL Molecular Graphics System, Httppymol Org., 2002.
[26] S.K. Lee, G.S. Chang, I.H. Lee, J.E. Chung, K.Y. Sung, K.T. No, The PreADME: pc-
based program for batch prediction of adme properties, EuroQSAR 9 (2004) 5–10.
[27] S.K. Lee, I.H. Lee, H.J. Kim, G.S. Chang, J.E. Chung, K.T. No, The PreADME
Approach: web-based program for rapid prediction of physico-chemical, drug
absorption and drug-like properties, EuroQSAR 2002 Des, Drugs Crop Prot. Process.
Probl. Solut. (2003) 418–420.
A. Belhassan et al. Heliyon 5 (2019) e02446
7

















