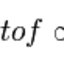Ab initio protein-protein docking algorithms often rely on experimental data to identify the most likely complex structure. We integrated protein-protein docking with the experimental data of chemical cross-linking followed by mass spectrometry. We tested our approach using 19 cases that resulted from an exhaustive search of the Protein Data Bank for protein complexes with cross-links identified in our experiments. We implemented cross-links as constraints based on Euclidean distance or void-volume distance. For most test cases the rank of the top-scoring near-native prediction was improved by at least two fold compared with docking without the cross-link information, and the success rate for the top 5 predictions nearly tripled. Our results demonstrate the delicate balance between retaining correct predictions and eliminating false positives. Several test cases had multiple components with distinct interfaces, and we present an approach for assigning cross-links to the interfaces. Employing the symmetry information for these cases further improved the performance of complex structure prediction.