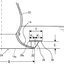
Michael Ashburner* and J. Jose Bonnert Department of Biochemistry and Biophysics University of California, San Francisco San Francisco, California 94143 During the normal development of the larval salivary gland of Drosophila melanogaster, considerable changes occur in the patterns of puffing activity. These can be seen as changes in the puffs of the gland’s polytene chromosomes, and occur as a con- sequence of changes in the titer of the insect’s growth and moulting hormone, ecdysone (for review see Ash- burner and Richards, 1976). In addition to the changes in gene activity normal to development, there are changes in the activities of a set of genes that occur as a direct consequence of subjecting animals to a wide variety of experimental insults, for example, a brief heat shock. The discovery of the induction of a unique set of puffs by heat shock (Ritossa, 1962) has led the way to an analysis of gene function and structure in Drosophila that is, so far, unique. The cytological facts can be summarized briefly (Ritossa, 1962, 1963, 1964a; Berendes and Holt, 1964; Berendes, Van Breugel and Holt, 1965; van Breugel, 1966; Ashburner, 1970; Ellgaard, 1972; Lewis, Helmsing and Ashburner, 1975). If Drosophila larvae or their excised tissues are subjected to a brief heat shock (for example, 40 min at 37”C, the normal culture temperature being 25”C), puffs are induced at a few specific sites (Figure 1). In D. melanogaster there are nine heat-inducible puffs, (33B, 63C, 64F, 678, 70A. 87A, 87C, 93D and 95D); in D. hydei there are six (32A, 36A, 48BC and 81 B; 31 C and 858 are small and variable in their response). The in vivo induction of the puffs by the heat shock is very rapid; it occurs within 1 min of the temperature increase although the puffs continue to increase in size for some 30-40 min (at 37°C) before regressing. The maximum sizes of the induced puffs are a function of the severity of the temperature shock, at least until lethal temperatures are met (Figure 2). The induction requires RNA, but not protein synthesis. In the ab- sence of protein synthesis, however, the induced puffs fail to regress unless the temperature is returned to normal (puff 48BC of D. hydei is an exception; Leen- ders and Beckers, 1972). Prolonged (for example, more than 1 hr) temperature shock results in addi- tional changes in puffing activity; most remarkable is the fact that all other puffs, puffs active at the time the temperature shock began, regress. It was the discovery that heat shock also results in the induction of the synthesis of a set of polypeptides ’ Permanent