Content uploaded by Matthias Kretzler
Author content
All content in this area was uploaded by Matthias Kretzler on Jan 25, 2024
Content may be subject to copyright.
Vol.:(0123456789)
Pediatric Nephrology
https://doi.org/10.1007/s00467-023-06256-7
REVIEW
Pediatric contributions andlessons learned fromtheNEPTUNE cohort
study
ZubinJ.Modi1,2· YanZhai1· JenniferYee1· HaileyDesmond1· WeiHao3· MatthewG.Sampson4,5,6,7·
ChristineB.Sethna8· Chia‑shiWang9· DebbieS.Gipson1· HowardTrachtman1 · MatthiasKretzler10,11· for the
NEPTUNE investigators
Received: 16 October 2023 / Revised: 20 November 2023 / Accepted: 5 December 2023
© The Author(s), under exclusive licence to International Pediatric Nephrology Association 2024
Abstract
Primary glomerular diseases are rare entities. This has hampered efforts to better understand the underlying pathobiology
and to develop novel safe and effective therapies. NEPTUNE is a rare disease network that is focused on patients of all ages
with minimal change disease, focal segmental glomerulosclerosis, and membranous nephropathy. It is a longitudinal cohort
study that collects detailed demographic, clinical, histopathologic, genomic, transcriptomic, and metabolomic data. The
goal is to develop a molecular classification for these disorders that supersedes the traditional pathological features-based
schema. Pediatric patients are important contributors to this ongoing project. In this review, we provide a snapshot of the
children and adolescents enrolled in NEPTUNE and summarize some key observations that have been made based on the
data accumulated during the study. In addition, we describe the development of NEPTUNE Match, a program that aims to
leverage the multi-scalar information gathered for each individual patient to provide guidance about potential clinical trial
participation based on the molecular characterization and non-invasive biomarker profile. This represents the first organized
effort to apply principles of precision medicine to the treatment of patients with primary glomerular disease. NEPTUNE has
proven to be an invaluable asset in the study of glomerular diseases in patients of all ages including children and adolescents.
Keywords Glomerular disease· Nephrotic syndrome· Pediatrics· Kidney biopsy· Biomarker· Precision medicine
Introduction
Glomerular diseases represent an important cause of chronic
kidney disease (CKD) in pediatric patients [1]. In most
regional registries of pediatric kidney disease around the
world, these disorders account for nearly a third of children
and adolescents with progressive loss of kidney function
leading to kidney failure and the eventual need for kidney
replacement therapy [2]. However, each glomerular disease
sub-type is a rare disease which has limited clinical and
basic science investigations of these disorders [3]. Conse-
quently, there has been little progress in the diagnosis and
Matthias Kretzler is the senior author.
* Howard Trachtman
howardtrachtman21@gmail.com
1 Division ofPediatric Nephrology, Department ofPediatrics,
University ofMichigan, AnnArbor, MI, USA
2 Susan B. Meister Child Health Research andEvaluation
Center, Department ofPediatrics, University ofMichigan,
AnnArbor, MI, USA
3 Department ofBiostatistics, University ofMichigan,
AnnArbor, MI, USA
4 Division ofPediatric Nephrology, Boston Children’s
Hospital, Boston, MA, USA
5 Harvard Medical School, Boston, MA, USA
6 Kidney Disease Initiative andMedical Population Genetics
Groups, Broad Institute, Cambridge, MA, USA
7 Division ofKidney Medicine, Brigham andWomen’s
Hospital, Boston, MA, USA
8 Cohen Children’s Medical Center, NewHydePark, NY, USA
9 Emory University School ofMedicine andChildren’s
Healthcare ofAtlanta, Atlanta, GA, USA
10 Department ofInternal Medicine, Division ofNephrology,
University ofMichigan, AnnArbor, MI, USA
11 Department ofComputational Medicine andBioinformatics,
University ofMichigan, AnnArbor, MI, USA
Pediatric Nephrology
treatment to improve health outcomes in pediatric patients
with glomerular diseases. In this review, we summarize the
design and accomplishments of the Nephrotic Syndrome
Study Network (NEPTUNE) and highlight how it has fos-
tered a better understanding of the molecular basis of pri-
mary glomerular disorders and facilitated the discovery of
targeted therapies in pediatric patients.
Primary glomerular diseases inpediatrics: brief
overview andgaps inknowledge
Nephrotic syndrome (NS) is a rare condition characterized
by proteinuria, hypoalbuminemia, and edema. It occurs in
patients of all ages and origins. The incidence of new-onset
disease generally ranges from 1 to 5 cases/100,000 pediatric
population per year. There are variations based on coun-
try, race, ethnicity, and social determinants of health with
higher incidence rates in select Asian countries [4]. It can
present with asymptomatic findings on urinalysis testing or
with overt clinical manifestations. In the minority of cases,
NS can be accompanied by hematuria, hypertension, and
reduced estimated glomerular filtration rate (eGFR).
The main glomerular disorders that cause NS in child-
hood are minimal change disease (MCD) and focal segmen-
tal glomerulosclerosis (FSGS) with membranous nephropa-
thy (MN) being far less common compared to adults [5].
Based on the high likelihood of MCD, the standard of care
for most children with new-onset NS is empirical treatment
with corticosteroids which are effective in most patients.
However, children can suffer relapsing disease. Along with
patients who are resistant to corticosteroids or who have
causes of NS other than MCD, they are candidates for ther-
apy with second-line immunosuppressive agents such as
calcineurin inhibitors, antimetabolites, and B-cell directed
therapies [6].
Recent scientific discovery has revealed that each of the
standard histopathological entities represents a heterogene-
ous cluster of subtypes characterized by distinct molecular
mechanisms of injury and disease progression [7]. There are
no non-invasive biomarkers that can identify the cause or
subtype of NS, predict the clinical course and long-term out-
come, or guide therapy. There are substantial gaps in knowl-
edge that have hindered the ability to advance understanding
of the natural history, optimal therapy, response to treatment,
and long-term health outcomes for individual children and
adolescents suffering from these disorders.
Objectives ofNEPTUNE
The Nephrotic Syndrome Study Network (NEPTUNE) is a
multicenter observational cohort study that has been sup-
ported for nearly 15years by the National Center of Advanc-
ing Translational Science and the National Institutes of
Health-National Institute of Diabetes, Digestive, and Kidney
Diseases [8]. The geographical distribution of participat-
ing sites is depicted in Fig.1 with an indication of those
that enroll pediatric patients. The objective of this rare dis-
ease network is to assemble a cohort of patients with MCD,
FSGS, and MN and, using a multi-scalar approach, collect
deep phenotypic information from each participant including
demographic, clinical, laboratory, genomic, transcriptomic,
and metabolomic data that can be analyzed with advanced
machine learning algorithms. The long-term goal is to gain
a more precise understanding of the underlying cause of
disease to implement the most appropriate individualized
treatment at the optimal time, i.e., use precision medicine to
treat patients with glomerular diseases (Fig.2). An exam-
ple of this integrative approach adopted by NEPTUNE is
the delineation of a subgroup of patients with NS in whom
tumor necrosis factor signaling was identified as a key path-
way promoting kidney injury [9].
The science and clinical applications of NEPTUNE are
based on the investigation of kidney tissues obtained at the
time of a diagnostic biopsy, a routine clinical procedure
in children and adults with proteinuria. However, pediat-
ric patients with new-onset NS are usually treated initially
without a diagnostic kidney biopsy [10]. Kidney biopsies
are only performed on children who do not achieve remis-
sion of proteinuria in response to therapy. This creates an
unintentional bias against enrollment of pediatric patients
close to the time of the clinical onset of disease. As a result,
the NEPTUNE protocol was modified to include enroll-
ment of pediatric patients within 30days of diagnosis of
NS, independent of a diagnostic kidney biopsy. We provide
a summary of NEPTUNE procedures and the experience of
pediatric participants, with or without a kidney biopsy, at
the time of enrollment.
Overview ofNEPTUNE
Clinical procedures
Since its inception in 2009, NEPTUNE has been recruiting
pediatric and adult NS patients into cohort A. It enrolled both
incident or prevalent adults and pediatric patients. Inclu-
sion criteria were (1) a protein:creatinine ratio (UPCR) > 0.5
(mg:mg) to capture the full range of the glomerular diseases,
(2) a clinically indicated kidney biopsy, and (3) histopatho-
logic confirmation of one of the three relevant diagnoses:
MCD, FSGS, and MN [8]. The UPCR threshold was raised
to 1.5 after the first 5-year funding period to increase the
likelihood of detecting active disease. The glomerular dis-
ease diagnosis was established by pathologists at the enroll-
ing site and confirmed by central review. Columbia scor-
ing and innovative measurements were performed based on
Pediatric Nephrology
review of biopsy material and digital analysis of whole slide
images [11, 12].
Considering the prevailing practice to treat children
with new-onset NS with corticosteroids prior to perform-
ing a kidney biopsy, in the second funding period, cohort
B was established to include pediatric patients with newly
diagnosed NS without a kidney biopsy. Cohort B enables
enrollment of participants with childhood NS as close as
possible to the disease onset. The eligibility criteria of
cohort B include (1) < 30 days of treatment for NS, (2)
age < 19years, (3) proteinuria/nephrotic defined as urinalysis
with > 2 + protein AND edema OR urinalysis with > 2 + pro-
tein AND serum albumin < 3 g/dL OR UPCR > 2 AND
serum albumin < 3g/dL. Cohort B is intended to maximize
early entry and collection of biosamples prior to prolonged
immunosuppressive medication. While entry into cohort A
required biopsy confirmation of primary glomerular disease,
inclusion in cohort B was based on a clinical assessment
indicating the presence of NS. During the third funding
period, Alport syndrome was added as a disease of inter-
est in NEPTUNE, and clinical and laboratory data will be
reported as the follow-up period is extended. IgA nephropa-
thy has not been included as a subgroup. Major exclusion
criteria are (1) kidney failure defined as dialysis or kidney
transplant, (2) sustained eGFR < 30 ml/min/1.73 m2, (3)
known secondary NS, and (4) inability to follow the study
protocol. Informed consent is obtained from theparent/legal
guardian and assent from children or adolescentsas appro-
priate. BP standards for participants in NEPTUNE are the
same as those for individuals who do not have glomerular
disease. Participant identification numbers are assigned at
enrollment or baseline visit. Follow-up visits are in person
at week 6, months 4 and 12 and then every 6months until
study termination. The overall features of cohorts A and B,
namely study visit schedule and procedures, are summarized
in Fig.3. The detailed procedures of cohort A have been
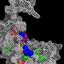
Fig. 1 Sites with pediatric enrollment in NEPTUNE. NEPTUNE pro-
vides a collaborative, investigational infrastructure across 32 North
American sites for translational research in focal segmental glomeru-
losclerosis (FSGS), minimal change disease (MCD), membranous
nephropathy (MN), pediatric non-biopsy cohort, Alport syndrome
Pediatric Nephrology
described previously [8]. For participants in cohort B, clini-
cal, demographic, physical examination, kidney pathology,
and laboratory data are obtained from electronic medical
records, while patient-reported information and quality of
life outcomes are collected using structured questionnaires,
for participants in both cohorts A and B.
Samples are processed in a central laboratory to assess
serum and urine creatinine and albumin to ensure data qual-
ity. Other laboratory results obtained include CBC, com-
plete metabolic panel, urinalysis, inflammatory markers
(C-reactive protein), serology, and virology. Home urinary
protein and disease status monitoring are done using Chem-
stixTM (Roche) urine dipstick and a short message service
(SMS) collecting information about edema status, adher-
ence to therapy, and school/work absences related to NS.
Patients who have a clinically indicated kidney biopsy after
enrollment into cohort B provide informed consent before
participating in biopsy-related biospecimen collection and
intra-renal gene expression profiling as in cohort A [8].
Biosamples including DNA, blood/plasma, and urine are
obtained and stored in the NIDDK Biosample Repository.
Data andanalytic methods
NEPTUNE uses the web-based ArborLink system (Arbor
Research, Ann Arbor, MI) as the data entry platform and
the data management system to create a fully relational
clinical research database with built-in features for detailed
reporting and auditing. The two primary outcomes meas-
ured are (1) event rate of change in urinary proteinu-
ria excretion (remission status) and (2) rate of change in
kidney function (decline of estimated GFR compared to
baseline). For estimation of GFR, the CKD-EPI equation is
used for patients ages ≥ 18years and modified Schwartz for
those < 18years. Baseline clinical characteristics, pathology
descriptors, laboratory data, and biomarkers are examined
in bivariate association tests between groups. Multivariate
regression models are used to identify associations between
clinical factors and outcomes and to compare time to event.
At the molecular level, whole genome sequencing (WGS)
has been performed followed by targeted analysis of 71 genes
implicated in NS to identify patients with putative monogenic
forms of NS or variants of kidney significance. This includes
APOL1 high risk (HR) genotype, which is an established risk
factor for kidney failure for those with African ancestry, and
steroid sensitive NS (SSNS) risk alleles, which have been
linked to immune-mediated pediatric disorders [13–15]. Kidney
transcriptional and signaling pathway mapping is performed
by examining nephron segment specific and single nuclear
gene expression profiles from an additional kidney biopsy core
obtained for research. Comprehensive urine/serum proteomic/
metabolomics signatures using a 2D-LCMS and a targeted pro-
teomic approach are integrated into NEPTUNE protocols to
identify non-invasive biomarkers. Other molecular signatures
including immune profiling were derived from single nuclear
RNA sequencing (RNAseq) from urine, peripheral blood mon-
onuclear cell (PBMC) and kidney tissue, when available.
Pediatric participation inNEPTUNE: biopsied
andnon‑biopsied cohorts
Since its inception, 823 patients have been enrolled in the NEP-
TUNE observational cohort study. Overall, the mean age is
27years and 58% are male. The racial breakdown is 53% White,
Fig. 2 Overview of NEPTUNE
objectives to achieve precision
medicine in nephrotic syndrome
Pediatric Nephrology
24% Black, 12% Asian, 5% multiracial, and 6% other. Patients of
Hispanic ethnicity account for 21% of the cohort. The primary
diagnosis is 26% MCD, 26% FSGS, 15% MN, 19% nephrotic
syndrome not otherwise specified, and 15% other glomerular
disease. At entry into the study, the mean baseline eGFR was
87ml/min/1.73 m2 (SD: 43.3), and meanUPCR was 4.52 (SD:
7.2)(mg protein:mg creatinine). The mean follow-up period
is32months (SD: 20).
The distribution of diagnosis and the age of the patients
enrolled in NEPTUNE are illustrated in Fig.4, confirming
the important contribution of pediatrics. Overall, 402 pedi-
atric patients have been enrolled in NEPTUNE, nearly 50%
of the total cohort, of whom 222 were in cohort A (biopsied)
and 180 in cohort B (non-biopsied). The pediatric contri-
bution to each primary diagnosis group is as follows: 99%
of the NS not otherwise specified, MCD 62%, FSGS 36%,
Fig. 3 Design of cohorts A
and B. Baseline and follow-
up sample and data collection
protocols for cohorts A and B.
NS, nephrotic syndrome; PE,
physical examination; SMS,
short messaging service; UPCR;
urine protein:creatinine ratio
Pediatric Nephrology
and MN 4%. The pediatric participants in NEPTUNE were
enrolled at 18 sites. Other than a younger age of children
enrolled in cohort B and the greater use of immunosuppres-
sive therapies in addition to corticosteroids in those enrolled
in cohort A, the two groups of patients are similar (Table1).
This suggests that investigations into the pathophysiology
of childhood NS will be generalizable to the full cohort. Of
note, although there is no lower age limit as an eligibility
criterion, no children with congenital nephrotic syndrome
have been enrolled in the study (Fig.5).
The standardized protocol that is followed in NEPTUNE
has enabled systematic evaluation of thetrajectory of key
clinical variables over the course of longitudinal follow-up
including body mass index (BMI), blood pressure (BP),
proteinuria, and eGFR. It is encouraging to note that most
of these factors are either stable or improve over time with
no evidence of increase in body weight or hypertension.
There is only a minimal increase in the number of pediatric
patients with CKD stage 3 or worse.
A central component of the NEPTUNE scientific effort is
the ancillary studies program that is designed to leverage the
detailed database and comprehensive biosample repository
that have been curated from participants. There have been
over 162 approved ancillary studies of which 100 (completed
or active) focus on or include pediatric participants cutting
across multiple research areas. These include epidemiology,
n = 29; transcriptomics, n = 23; biomarkers, n = 20; histopa-
thology, n = 14; and genetics, n = 14.
Pediatrics lessons fromNEPTUNE
Prognosis There is uncertainty whether the prognosis of
FSGS is better or worse in pediatric versus adult patients
with FSGS. In order to address this concern, a study was
conducted using pooled and parallel analyses from three
complementary data sources: (1) NEPTUNE, n = 166;
(2) FSGS-CT, a multicenter US/Canada clinical trial that
included steroid resistant FSGS patients randomized to
cyclosporine vs. dexamethasone + mycophenolate, n = 132;
and (3) Kidney Research Network (KRN), a multicenter US
electronic health record-based registry from academic and
community nephrology practices, n = 184 [16]. The primary
outcomes were time to kidney failure, composite of kidney
failure or 40% decline in eGFR and complete and/or par-
tial remission of proteinuria. The patients were divided into
three age groups, children < 13years (n = 127), adolescents
13–17years (n = 102), and adults ≥ 18years (n = 253). The
combined cohort included 215 (45%) females and 138 (29%)
participants who reported as Black race. Overall, the median
time to kidney failure was 11.9years (IQR 5.2, 19.1years)
with age group comparisons children vs. adults HR = 1.12
[95% CI 0.83 to 1.52], adolescents vs. adults HR = 1.06 [95%
CI 0.75 to 1.50]. The median time to the composite end-
point was 5.7years (IQR 1.6, 15.2years) with hazard ratio
estimates for children vs. adults [HR = 1.12 [95% CI 0.83
to 1.52]]; adolescents vs. adults [HR = 1.06 [95% CI 0.75
to 1.50]]. The results were unchanged after adjustment for
sex, disease duration, APOL1 risk genotype, initial level of
proteinuria, and treatment. In pooled analysis, adults had
an estimated eGFR slope of − 1.71ml/year (95% CI − 3.23
to − 0.19), adolescents − 3.84 (95% CI − 5.86 to − 1.82), and
children − 3.32 (95% CI − 5.13 to − 1.51). Children and ado-
lescents had higher starting eGFR than adults, but there was
no statistically significant difference in the rate of decline in
eGFR over time. After 12months of follow-up, 49% (95%
CI 30 to 69%) of children, 38% (95% CI 20 to 57%) of ado-
lescents, and 25% (95% CI 16 to 34%) of adults had reached
complete remission. In addition, there was no difference in
time to the composite of a complete remission or achiev-
ing the novel FSGS partial remission endpoint, namely 40%
reduction in UPCR to < 1.5 by age. Thus, the natural his-
tory and kidney health outcomes of FSGS are comparable
across the lifespan and adverse consequences of this glo-
merular disease fall equally on pediatric and adult patients.
This provides strong justification for inclusion of children
and adolescents with adults in clinical trials of novel thera-
pies for FSGS. Work done within the pediatric participants
enrolled in NEPTUNE demonstrated that urinary excretion
Fig. 4 Distribution of the
overall NEPTUNE cohort by
diagnosis and age. A Circle plot
of diagnoses — focal segmental
glomerulosclerosis (FSGS),
minimal change disease (MCD),
membranous nephropathy
(MN), Alport syndrome, and
no diagnosis. B Circle plot of
showing distribution by age of
the NEPTUNE cohort
Pediatric Nephrology
Table 1 Clinical and demographic characteristics of pediatric participants in NEPTUNE
a Values were consolidated between central and local testing with preference given to central results
b IQR reflects the size of the range
BMI body mass index, IQR interquartile range, IST immunosuppressive therapy, RAAS renin-angiotensin-aldosterone system, SD standard devia-
tion, UPCR urine protein: creatinine ratio
Overall
(n = 402)
Biopsy cohort A
(n = 222)
Non-biopsy cohort B
(n = 180)
Age, years, mean (SD) 7.6 (5.0) 9.8 (5.0) 4.8 (3.5)
Follow up time, months, median 30.00 37.00 20.00
Biologic sex, N = 398, n (%)
Female 177 (44.47%) 99 (44.80%) 78 (44.07%)
Race, N = 401, n (%)
Asian 39 (9.73%) 20 (9.05%) 19 (10.56%)
Black or African American 111 (27.68%) 73 (33.03%) 38 (21.11%)
Native American/Alaskan Native/First Nation 4 (1.00%) 0 (0.00%) 4 (2.22%)
Native Hawaiian/Other Pacific Islander 2 (0.50%) 1 (0.45%) 1 (0.56%)
White 186 (46.38%) 97 (43.89%) 89 (49.44%)
Multi-racial 34 (8.48%) 19 (8.60%) 15 (8.33%)
Unknown 25 (6.23%) 11 (4.98%) 14 (7.78%)
Hispanic, N = 401, n (%) 81(20.20%) 45 (20.36%) 36 (20.00%)
Number of APOL1 risk alleles, N = 236, n (%)
0 172 (72.88%) 129 (70.49%) 43 (81.13%)
1 31 (13.14%) 22 (12.02%) 9 (16.98%)
2 33 (13.98%) 32 (17.49%) 1 (1.89%)
Clinical status at baseline visit
BMI, N = 360, n (%)
Underweight 10 (2.78%) 6 (2.93%) 4 (2.58%)
Normal 152 (42.22%) 77 (37.56%) 75 (48.39%)
Overweight 81 (22.50%) 46 (22.44%) 35 (22.58%)
Obese 117 (32.50%) 76 (37.07%) 41 (26.45%)
Hypertension, N = 368, n (%)
Normal 129 (35.05%) 87 (41.63%) 42 (26.42%)
Elevated 51 (13.86%) 29 (13.88%) 22 (13.84%)
Stage 1 138 (37.50%) 68 (32.54%) 70 (44.03%)
Stage 2 50 (13.59%) 25 (11.96%) 25 (15.72%)
eGFR, mL/min/1.73 m2, N = 380, median (IQRb) 95.61 (39.36) 93.77 (33.93) 98.97 (50.89)
eGFR ≤ 60, n (%) 21 (5.53%) 15 (6.98%) 6 (3.64%)
eGFR (60,120), n (%) 269 (70.79%) 165 (76.74%) 104 (63.03%)
eGFR > 120, n (%) 90 (23.68%) 35 (16.28%) 55 (33.33%)
UPCR, mg/mg, N = 365, median (IQRb) 1.38 (6.44) 1.18 (4.77) 2.00 (11.95)
UPCR (0,0.3), n (%) 118 (32.33%) 55 (26.83%) 63 (39.38%)
UPCR (0.3,1.0), n (%) 50 (13.70%) 39 (19.02%) 11 (6.88%)
UPCR (1.0,3.0), n (%) 51 (13.97%) 42 (20.49%) 9 (5.63%)
UPCR > 3.0, n (%) 146 (40.00%) 69 (33.66%) 77 (48.13%)
Serum albumin, g/dL, N = 320, mean (SD)a3.31 (0.98) 3.42 (1.01) 3.12 (0.90)
Total cholesterol, mg/dL, N = 304, mean (SD)a318.53 (138.16) 298.38 (136.29) 351.64 (135.35)
Any edema, N = 393, n (%)
Absent 234 (59.54%) 129 (60.56%) 105 (58.33%)
Present 159 (40.46%) 84 (39.44%) 75 (41.67%)
Prior medication use, n (%) multiple possible
Steroids (days of prior) 340 (84.58%) 166 (74.77%) 174 (96.67%)
Calcineurin inhibitor 76 (18.91%) 73 (32.88%) 3 (1.67%)
Mycophenolate mofetil 33 (8.21%) 32 (14.41%) 1 (0.56%)
Cyclophosphamide 19 (4.73%) 19 (8.56%) 0 (0.00%)
Rituximab 3 (0.75%) 3 (1.35%) 0 (0.00%)
Other IST 0 (0.00%) 0 (0.00%) 0 (0.00%)
RAAS blockade 165 (41.04%) 119 (53.60%) 46 (25.56%)
Pediatric Nephrology
of epidermal growth factor may be a useful non-invasive
prognostic biomarker, with higher levels at baseline being
associated with a slower rate of eGFR decline during fol-
low-up [17]. This is an example of the translational science
emerging from NEPTUNE that hopefully will improve the
management of NS in children.
Cardiovascular risk Patients with primary proteinuric glo-
merulopathies are at increased risk for adverse cardiovas-
cular (CV) outcomes. However, the effects of these CV risk
factors on disease outcomes in pediatric patients versus
adults with primary proteinuric glomerulopathies have not
been well-characterized.
To uncover the relationship between hypertension and
blood pressure variability (BPV) with kidney outcomes
in patients with primary glomerulopathies, studies were
conducted in the NEPTUNE cohort [18]. Hypertensive BP
status was defined as previous history of hypertension or
BP ≥ 140/90mm Hg for adults, ≥ 95th percentile for children
at baseline. BPV was evaluated using average real variability
(ARV), measuring differences in BP between consecutive
visits and standard deviation (SD) reflecting overall vari-
ability. Adjusted regression analysis was used to evaluate
the association between hypertensive BP status and BPV
between visits with clinical outcomes of complete remission
of proteinuria, composite of kidney failure or 40% decline in
eGFR, and eGFR slope [18].
The study population included 443 participants with
baseline BP measurements recorded, including 147 children
(< 18years) and 296 adults (≥ 18years). At baseline, 261
of the participants (59%) were previously diagnosed with
hypertension (n = 209) or had a hypertensive BP level at
baseline (n = 54). Hypertension was more common in adults
than children, but children were more likely to be diagnosed
with uncontrolled hypertensive BP at baseline. In fully
adjusted models, the underlying disease cohort was signifi-
cantly associated with hypertension in adults (p = 0.036) but
not in children (p = 0.9). The odds of hypertension were 5.5
times greater in patients with IgA nephropathy (p = 0.005)
and 3.8 times greater in patients with FSGS (p = 0.01)
compared with MCD. In adults, hypertension was associ-
ated with a lower risk of complete remission (HR = 0.48,
Fig. 5 NEPTUNE Match. A Figure illustrating the overall structure of NEPTUNE Match. B Schematic summarizing the communication compo-
nent of the Match program for each participant
Pediatric Nephrology
p < 0.001). Baseline hypertension was not associated with
eGFR slope. In children, hypertension was not associated
with complete remission, the composite end point, or eGFR
slope. Higher systolic and diastolic ARV were associated
with a lower likelihood of complete remission ever (SBP
ARV: HR = 0.92, p = 0.01 and DBP ARV: HR = 0.95,
p = 0.041). These findings demonstrated that hypertension
is associated with worse clinical outcomes in adults com-
pared to children, but that greater BPV is linked to poor
kidney outcomes across the lifespan in patients with primary
proteinuric glomerulopathies.
To explore the association between obesity and primary
glomerulopathy and its progression, data was collected
from patients enrolled in NEPTUNE, including 360 adults
(≥ 18years) and 181 children (< 18years). The primary
outcomes were kidney failure (defined as eGFR < 15mL/
min/1.73 m2 at two consecutive visits) or a diagnosis of kid-
ney failure, CKD progression (defined as having both an
eGFR decline by 40% and an eGFR < 90mL/min/1.73 m2,
or kidney failure), and complete remission ever (defined as
a UPCR < 0.3g/g at any time point after diagnosis) [19].
Among adult patients, 156 (43%) were obese compared to
68 (38%) of the pediatric patients. In adults, adjusted linear
and logistic models indicated that obesity was associated
with higher triglycerides (β = 41.92, 95% CI 17.12, 66.71,
p = 0.001), higher systolic BP (β = 6.49, 95% CI 2.41, 10.56,
p = 0.002), increased odds of dyslipidemia (OR = 1.74, 95%
CI 1.30, 2.32, p < 0.001), and lower HDL (β = − 6.92, 95%
CI − 9.32, − 4.51, p < 0.001). In children, adjusted models
showed a significant association between obesity and higher
systolic BP index (β = 0.04, 95% CI 0.02, 0.06, p < 0.001)
and hypertension (OR = 1.43, 95% CI 1.04, 1.98, p = 0.03).
These results demonstrate that obesity in both adults and
children with primary glomerulopathies is linked to adverse
CV outcomes, even though there were no significant associa-
tions between obesity and kidney failure or the progression
of CKD. Meanwhile during follow-up, 274 (74%) adults and
144 (75%) children reached complete or partial remission of
proteinuria. Obesity was associated with a lower prospect of
ever reaching complete remission in adults (HR = 0.80, 95%
CI 0.69, 0.88, p < 0.001) and children (HR = 0.72, 95% CI
0.61, 0.84, p < 0.001). These data demonstrate the impor-
tance of therapeutically targeting obesity to improve long-
term clinical outcomes in patients with primary proteinuric
glomerulopathies.
To explore the association between smoking exposure
and CV and kidney outcomes in the subset of patients with
primary glomerulopathies, data were collected from patients
enrolled in NEPTUNE, including 371 adults and 192 chil-
dren. The primary outcomes were UPCR and eGFR as well
as complete remission, CKD progression, and kidney fail-
ure as defined above. Secondary outcomes included CV dis-
ease diagnoses such as coronary artery disease, stroke, heart
failure, peripheral vascular disease, thromboembolic events,
and cancer diagnosis [20].
The adult sub-cohort comprised 191 (52%) non-smokers,
54 (15%) active smokers, 108 (29%) former smokers, and
18 (5%) passive smokers. The cohort of children consisted
of 156 (81%) non-smokers and 32 (17%) passive smokers.
In adults, active smoking was significantly associated with
greater total cholesterol (β = 17.91, 95% CI 0.06, 35.76,
p = 0.049), and former smokers had a higher frequency of
thromboembolic events (9.3%) compared to other tobacco
exposure groups. In the pediatric cohort, passive smoke
exposure was significantly associated with increased UPCR
(β = 1.23, 95% CI 0.13, 2.33, p = 0.03). Future research will
be needed to explore the impact of passive smoking and
potential confounding factors on the course of these glomer-
ular diseases. Due to the adverse effects of tobacco exposure
on cholesterol levels in adults and proteinuria in children,
strategies should be implemented to reduce smoke exposure,
such as counseling for caregivers and family members of
patients with primary glomerulopathies.
Genetics The NEPTUNE study has provided important
insight into the incidence of monogenic causes of nephrotic
syndrome in pediatric patients. Previous reports suggested
that up to 30% of children with steroid-resistant NS have
a causative genetic mutation [21]. Using a similar patho-
genicity filtering strategy, only 5.6% of pediatric NEP-
TUNE patients who did not achieve complete remission
had a putative monogenic form of their condition [22]. The
lower incidence of monogenic disease in NEPTUNE versus
other cohorts may be related to more stringent criteria that
were applied to variant calling. In addition, the collagen IV
genes were not tested in this study. Given our understand-
ing of the prevalence of pathogenic COL4A3/4/5 variants
in pediatric SRNS patients, in this pediatric SRNS subset,
we would predict no more than a doubling of prevalence.
Genetic studies in the NEPTUNE cohort have also helped
to elucidate the impact of Apolipoprotein L1 (APOL1) risk
variants on Black children with proteinuric kidney disease.
In a study of 48 NEPTUNE participants who were Black
(via self-report) and had childhood onset of disease, 56% had
low risk APOL1 genotypes (0 or 1 risk variant), and 44% had
a high-risk genotype (2 risk variants) [23]. Children with a
high-risk variant had a significantly increased age of onset
of disease (14 vs. 11years), a lower eGFR at baseline (74
vs. 94ml/min), and a more rapid decline of eGFR (− 13.4%
vs. − 3.2% per year). These findings were replicated in a
similar cohort of children in the CKiD study [24].
Beyond direct kidney phenotypes, there were links
between high-risk genotypes, poor birth outcomes, and
FSGS, further supporting the potential role of endothelial
dysfunction in APOL1 pathobiology. In two independent
studies, compared to those with a low-risk genotype, Black
Pediatric Nephrology
FSGS patients with a high-risk genotype had 3-fold and
9.6-fold increased odds of being born preterm [24, 25]. In
contrast, in a 2017 study of healthy populations, there was
no association between maternal and fetal APOL1 genotypes
and prematurity [23]. However, these studies were under-
powered to detect associations with other metrics of poor
birth outcomes. Subsequently, multiple independent stud-
ies discovered an association of APOL1 high-risk genotypes
with preeclampsia.
Home monitoring For most children, NS is a chronic,
relapsing-remitting condition that requires vigilant moni-
toring. Eighty percent of children initially responsive to
corticosteroid treatment will relapse at least once, and half
will relapse frequently or when treatments are tapered or
stopped [10]. The unpredictable disease relapses cause sig-
nificant uncertainty and stress for patients and families [26].
If relapses are not treated and controlled in a timely manner,
they can lead to severe edema and complications such as
acute kidney injury, thromboembolism, or serious infections
[10]. Effective NS management in children relies on home
urine protein monitoring using test strips. This practice can
detect disease relapses before symptoms are apparent to
patients and their families and enable physicians to initi-
ate or adjust treatments before complications occur. Most
relapses are treated with high-dose corticosteroids, which
are tapered as soon as remission is achieved in order to mini-
mize treatment side effects [10, 27]. Thus, monitoring for
proteinuria remission and close communication with physi-
cians when it occurs is also important. However, effective
methods to support families entrusted with home disease
monitoring are lacking. Suboptimal adherence with home
urine testing, confusion over what the test results mean, and
delayed communication with providers are frequent issues
limiting effective management in childhood NS.
To improve disease monitoring at home, NEPTUNE imple-
mented a text-messaging system (SMS) beginning in June 2015
among all study participants < 19years of age. Text messages
are sent to either the parent/guardian or adolescent participants
daily to weekly for 1year and solicit numerical responses to
questions on urine protein test results, the presence of edema,
missed school or work, recent health issues, and medication
adherence. Responses are analyzed and email alerts are sent
to study teams for new relapses, onset of edema, or missed
medication. In an analysis of 127 eligible NEPTUNE partici-
pants treated between June 2015 and March 2018, SMS was
well-accepted by patients and families and reliably captured
disease activity and symptoms [28]. Adoption of the mobile
technology was high; 119 out of 127 participants (94%) agreed
to SMS monitoring, and 112 (94%) continued to respond to
texts throughout the 1-year study duration. Respondents
had high engagement with the text messages, with a median
response of 87% (IQR 68–97%) to texts. Importantly, concord-
ance between patient-reported home urine protein results and
urinalyses performed in clinic was 94% (kappa = 0.88), and
concordance between patient-reported edema and physician-
documented edema on physical exam was 96% (kappa = 0.92).
Based on SMS reporting, a total of 108 relapse events were
detected compared to the 41 events captured by clinical vis-
its. With more frequent proteinuria data gathered via SMS, a
more precise estimated median time-to-remission of 22days
(IQR 13–64) was identified than when using clinical visit data
(50days, IQR 21–150). This experience in NEPTUNE pro-
vided much-needed evidence that mobile health tools are an
effective method to assist patients, families, and providers in NS
management. Further research is needed to determine whether
use of SMS-based or other mobile health communication strat-
egies will lead to improved health outcomes in children and
adolescents with NS (Table2).
NEPTUNE Match
The application of state-of-the-art approaches in NEPTUNE
to the investigation of primary glomerular diseases has
resulted in a mechanism-based classification of glomeru-
lar disease. Using this comprehensive dataset, NEPTUNE
is moving the needle toward precision medicine-based
Table 2 Summary of findings in key studies
Reference number Area of investigation Findings
[16] Prognosis of FSGS Rate of disease progression and likelihood of remission is comparable in children, adolescents,
and adults with FSGS
[18–20] Cardiovascular outcomes Hypertension is more impactful in adults
Obesity is associated with lower risk of disease remission
Passive smoking associated with increased proteinuria
[21–25, 29, 30] Genetics Only 5.6% of children with nephrotic syndrome have monogenic disease
High-risk APOL1 genotypes associated with more aggressive disease and adverse maternal-fetal
outcomes during pregnancy
[28] Clinical management Communication via text messaging is accepted by patients and detects more relapses and earlier
response to treatment
Pediatric Nephrology
therapeutics, namely implementing trials that will enable
the right medicine to be made available to the right patient
at the right time.
To achieve this goal, a program called NEPTUNE Match
has been designed and implemented [31]. It creates an infor-
mational bridge between the basic science discovery pipeline
and the initiation of targeted clinical trials. The components
include clinical and laboratory data, machine learning-based
integrative clinical outcome modeling, advanced digital kid-
ney histopathology tools, and systems genetics approaches.
Whole genome sequencing and expression studies from
micro-dissected kidney tissue compartments are linked.
Single-cell RNA sequencing and organoid technologies have
been developed and associated with the numerous existing
tissue level datasets.
NEPTUNE Match represents a collaborative effort
between the NIH-NIDDK-funded network and private
partners in the NEPTUNE Pre-competitive Public–Private
Partnership Platform (NP5). NP5 is a dynamic framework
to enable pre-competitive assessment of pathways targeted
in clinical trials, initiated by the private biotechnology
partners or independent academic investigators. In paral-
lel, NEPTUNE Match provides patients with cutting edge
information about the molecular features of their disease
for consideration when they weigh the pros and cons about
participation in available clinical trials.
In the initial step, a Molecular Nephrology Board reviews
the broad range of clinical, histopathological, and labora-
tory data, routine and experimental, for each patient. This
yields a molecular categorization based on the underlying
mechanism of disease, which supplants traditional diagnos-
tic categories. Pathway activation signatures targeted by
clinical trials pursued in glomerular diseases are reviewed
for each participant. Non-invasive biomarker profiles are
developed to allow molecular pathway classification for
individual patients. The pathway assessment can then be
accomplished in real time without requiring a kidney biopsy.
A Match report is generated that indicates the individual
pathway activation scores for a specific patient relative to
the overall NEPTUNE population and graphically depicts
the degree of fit between the molecular disease pathway(s)
and proposed clinical trials.
A protocol has been established in collaboration with
patient advocates and bioethicists to effectively communi-
cate the results of the NEPTUNE Match report to the patient
and the attending nephrologist to assist them in their eval-
uation and selection of available clinical trials. Sites that
are participating in NEPTUNE Match are trained on how
to convey the information contained in the Match report.
The communication team may include the local clinician-
investigator, study coordinator, and in addition the adult
patient/pediatric patient/decision-making proxy for minor
participants. This communication includes a review of
the trial matching assessment, time to address questions,
teach back to confirm comprehension, and assessment of
potential psychological distress and anxiety caused by the
experimental information. The psychological impact of
receiving this information is evaluated using an instrument
that was adapted from a tool used to assess the psychosocial
consequences of returning genomic findings to patients in
the research setting and clinical practice. It is important to
emphasize that decisions concerning clinical management
of the study participants as well as clinical trial enrollment
are made by the patient and their care provider and are not
part of NEPTUNE Match.
Conclusion
NEPTUNE represents an organized multidisciplinary
initiative to better understand primary glomerular dis-
eases and to develop precision medicine approaches for
the treatment of affected patients. It has expanded our
understanding of these disorders and has shed new light
on the disease course, management, and outcomes in
pediatric patients. Looking to the future, we will con-
tinue to push the program at the cutting edge of research
to capture the rapidly changing landscape of pediatric
NS across the USA and Canada. With the next genera-
tion technologies now able to define molecular programs
in individual cells andspatially defined compartments
within kidney biopsies, this deep tissue level understand-
ing of the disease process can be linked with the lived
experience of the patients to enable the implementation
of molecular anchored management strategies. NEP-
TUNE Match represents the first application of preci-
sion medicine in nephrology with the aim of developing
targeted therapies that will be more effective and have
fewer adverse effects for patients with primary glomeru-
lar disease. Expanding the scope of collaboration with
industry partners will help sustain the discovery pipe-
line needed to define individualized therapeutic targets
through associated biomarkers for patients with glo-
merular disease. In addition, it will expand the number
of therapeutic options available to patients and provide
them with a wider range of clinical trial options that
they can consider and select based on potential efficacy,
adverse effects, and feasibility. NEPTUNE Match offers
an opportunity to create a permanent infrastructure of
highly qualified sites that have the personnel, institu-
tional resources, and experiences required to conduct
trials of novel precision medicine-based therapies and
innovative trial strategies. Overall, NEPTUNE has
proven to be an invaluable asset in the study of glomer-
ular diseases in patients of all ages including children
and adolescents.
Pediatric Nephrology
Acknowledgements The authors wish to thank Hannah Strum for her
help in compiling the data from studies conducted in NEPTUNE and
Lalita Subramanian for her expert assistance in the preparation of the
manuscript.
Members of the Nephrotic Syndrome Study Network
(NEPTUNE)
NEPTUNE collaborating sites
Atrium Health Levine Children’s Hospital, Charlotte, SC: Susan
Massengill*, Layla Lo#
Cleveland Clinic, Cleveland, OH: Katherine Dell*, John O’Toole*,
John Sedor**, Blair Martin#
Children’s Hospital, Los Angeles, CA: Ian Macumber*, Silpa
Sharma#
Children’s Mercy Hospital, Kansas City, MO: Tarak Srivastava*,
Kelsey Markus#
Cohen Children’s Hospital, New Hyde Park, NY: Christine Sethna*,
Suzanne Vento#
Columbia University, New York, NY: Pietro Canetta*
Duke University Medical Center, Durham, NC: Opeyemi Olabisi*,
Rasheed Gbadegesin**, Maurice Smith#
Emory University, Atlanta, GA: Laurence Greenbaum*, Chia-shi
Wang*, Emily Yun#
The Lundquist Institute, Torrance, CA: Sharon Adler*, Janine
LaPage#
John H Stroger Cook County Hospital, Chicago, IL: Amatur
Amarah*, Mathew Itteera#
Johns Hopkins Medicine, Baltimore, MD: Meredith Atkinson*,
Miahje Williams#
Mayo Clinic, Rochester, MN: John Lieske, Marie Hogan, Fernando
Fervenza.
Medical University of South Carolina, Charleston, SC: David
Selewski*, Cheryl Alston#
Montefiore Medical Center, Bronx, NY: Kim Reidy*, Michael Ross*,
Frederick Kaskel**, Patricia Flynn#
New York University Medical Center, New York, NY: Laura Malaga-
Dieguez*, Olga Zhdanova**, Laura Jane Pehrson#, Melanie Miranda#
The Ohio State University College of Medicine, Columbus, OH:
Salem Almaani*, Laci Roberts#
Stanford University, Stanford, CA: Richard Lafayette*, Shiktij Dave#
Temple University, Philadelphia, PA: Iris Lee**
Texas Children’s Hospital at Baylor College of Medicine, Houston,
TX: Shweta Shah*, Sadaf Batla# #
University Health Network Toronto: Heather Reich*, Michelle
Hladunewich**, Paul Ling#, Martin Romano#
University of California at San Francisco, San Francisco, CA: Paul
Brakeman*
University of Colorado Anschutz Medical Campus, Aurora, CO:
James Dylewski* Nathan Rogers#
University of Kansas Medical Center, Kansas City, KS: Ellen
McCarthy*, Catherine Creed#
University of Miami, Miami, FL: Alessia Fornoni*, Miguel Bandes#
University of Michigan, Ann Arbor, MI: Matthias Kretzler*, Laura
Mariani*, Zubin Modi*, A Williams#, Roxy Ni#
University of Minnesota, Minneapolis, MN: Patrick Nachman*,
Michelle Rheault*, Amy Kowalski#, Nicolas Rauwolf#
University of North Carolina, Chapel Hill, NC: Vimal Derebail*,
Keisha Gibson*, Anne Froment#, Sara Kelley#
University of Pennsylvania, Philadelphia, PA: Lawrence Holzman*,
Kevin Meyers**, Krishna Kallem#, Aliya Edwards#
University of Texas San Antonio, San Antonio, TX: Samin Sharma**
University of Texas Southwestern, Dallas, TX: Elizabeth Roehm*,
Kamalanathan Sambandam**, Elizabeth Brown**, Jamie Hellewege.
University of Washington, Seattle, WA: Ashley Jefferson*, Sangeeta
Hingorani**, Katherine Tuttle**§, Linda Manahan#, Emily Pao#, Kelli
Kuykendall§
Wake Forest University Baptist Health, Winston-Salem, NC: Jen
Jar Lin**
Washington University in St. Louis, St. Louis, MO: Vikas
Dharnidharka*
Data analysis and coordinating center
University of Michigan: Matthias Kretzler*, Brenda Gillespie**,
Laura Mariani**, Zubin Modi**, Eloise Salmon**, Howard Trachtman**,
Tina Mainieri, Gabrielle Alter, Michael Arbit, Hailey Desmond, Sean
Eddy, Damian Fermin, Wenjun Ju, Maria Larkina, Chrysta Liencze-
wski, Rebecca Scherr, Jonathan Troost, Amanda Williams, Yan Zhai;
Arbor Collaborative for Health: Colleen Kincaid, Shengqian Li,
Shannon Li; Cleveland Clinic: Crystal Gadegbeku**; Duke Univer-
sity: Laura Barisoni**, John Sedor**; Harvard University: Matthew G
Sampson**; Northwestern University: Abigail Smith**; University of
Pennsylvania: Lawrence Holzman**, Jarcy Zee**
Digital pathology committee
Carmen Avila-Casado (University Health Network), Serena
Bagnasco (Johns Hopkins University), Lihong Bu (Mayo Clinic),
Shelley Caltharp (Emory University), Clarissa Cassol (Arkana),
Dawit Demeke (University of Michigan), Brenda Gillespie (University
of Michigan), Jared Hassler (Temple University), Leal Herlitz (Cleve-
land Clinic), Stephen Hewitt (National Cancer Institute), Jeff Hodgin
(University of Michigan), Danni Holanda (Arkana), Neeraja Kamb-
ham (Stanford University), Kevin Lemley, Laura Mariani (University of
Michigan), Nidia Messias (Washington University), Alexei Mikhailov
(Wake Forest), Vanessa Moreno (University of North Carolina),
Behzad Najafian (University of Washington), Matthew Palmer (Uni-
versity of Pennsylvania), Avi Rosenberg (Johns Hopkins University),
Virginie Royal (University of Montreal), Miroslav Sekulik (Columbia
University), Barry Stokes (Columbia University), David Thomas (Duke
University), Ming Wu (University of New York), Michifumi Yamashita
(Cedar Sinai), Hong Yin (Emory University), Jarcy Zee (University
of Pennsylvania), Yiqin Zuo (University of Miami). Co-Chairs: Laura
Barisoni (Duke University), Cynthia Nast (Cedar Sinai).*Principal
Investigator; **Co-investigator; #Study Coordinator; §Providence
Medical Research Center, Spokane, WA
Funding The Nephrotic Syndrome Study Network (NEPTUNE)
is part of theRare Diseases Clinical Research Network(RDCRN),
which is funded by the National Institutes of Health (NIH) and led by
theNational Center for Advancing Translational Sciences(NCATS)
through itsDivision of Rare Diseases Research Innovation (DRDRI).
NEPTUNE is funded under grant number U54DK083912 as a col-
laboration between NCATS and the National Institute of Diabetes
and Digestive and Kidney Diseases (NIDDK). Additional funding
and/or programmatic support is provided by the University of Michi-
gan, NephCure Kidney International, Alport Syndrome Foundation,
and the Halpin Foundation. RDCRN consortia are supported by the
RDCRN Data Management and Coordinating Center (DMCC), funded
by NCATS and the National Institute of Neurological Disorders and
Stroke (NINDS) under U2CTR002818. MGS is supported by NIH
R01DK119380, 2U54DK083912, RC2DK122397, and the Pura Vida
Kidney Foundation.
Data Availability Neptune clinical data are available to researchers
upon reasonable request to NEPTUNE-STUDY@umich.edu,and will
become available through the NIH repository.
Declarations
Conflict of interest Dr. Modi reports research funding outside of this
workfrom the National Institutes of Health, Centers for Disease Con-
trol, the Patient-Centered Outcomes Research Institute, Travere Thera-
peutics, and Boehringer Ingelheim. He is also the current director of
the Kidney Research Network Coordinating Center. Dr. Sampson is
Pediatric Nephrology
on the scientific advisory board of Natera. Dr Wang received research
support from the National Institute of Diabetes and Digestive and Kid-
ney Diseases, the Centers for Disease Control and Prevention, Neph-
Cure Kidney International (NKI), the European Society of Pediatric
Nephrology—Pediatric Nephrology Research Consortium, Boehring-
er-Ingelheim, IQVIA, and Genentech outside the submitted work.
Dr. Gipson reports past research funding from National Institutes of
Health, Centers for Disease Control, US Food and Drug Administra-
tion, Travere Therapeutics, Reata, Novartis, and Boehringer Ingel-
heim; past consulting fees paid to the University of Michigan form
Roche/Genentech and Vertex; past participation in Data Safety Moni-
toring Board for National Institutes of Health; past director of Kidney
Research Network Coordinating Center; and unpaid project co-lead of
the National Kidney Foundation improving vaccination in the kidney
disease community project, past co-lead of the Kidney Health Initia-
tive Pediatric IgA nephropathy project, past member of the Kidney
Health Initiative FSGS outcomes project, and past planning commit-
tee member for the NephCure and Kidney Health Initiative sponsored
workshop entitled Pathways to SGLT2i for renoprotection in pediatric
CKD. Dr. Trachtman reports employment with RenalStrategies LLC.
He has active consultancy agreements with Aclipse, Angion, Goldfinch
Bio, Maze Therapeutics, Natera (RenaSight), Otsuka (DSMB Chair for
Pediatric Trials), Travere Therapeutics, Inc., Boehringer-Ingelheim,
PhaseV and Walden; honoraria for participation in glomerular disease
panels organized by Astellas and Reata; Steering Committee and Sci-
entific Advisory Board for DUPRO (DUPLEX and POTECT Trials)
for Travere Therapeutics, Inc.; member of the Kidney Health Initiative
Board of Directors; Editorial board member of Pediatric Nephology,
Glomerular Diseases, and Kidney360; serves as a partner with Neph-
Cure Kidney International in efforts to promote pediatric participa-
tion in clinical trials for glomerular diseases (PIONEER). Dr. Kretzler
reports grants from NIH, non-financial support from University of
Michigan, funding through the University of Michigan from Goldfinch
Bio, Boehringer-Ingelheim, Certa, Travere and Maze therapeutics, as
well as grants and contracts outside the submitted work through the
University of Michigan with NIH, Chan Zuckerberg Initiative, JDRF,
Roche, Astra-Zeneca, NovoNordisk, Moderna, European Union Inno-
vative Medicine, Chinook, Angion Pharmaceuticals, RenalytixAI, Eli
Lilly, Gilead, Regeneron, IONIS, and Janssen; consulting fees through
the University of Michigan from Astellas, Poxel, Janssen, NovoNor-
disc; and serves on the NIH-NCATS council and the NephCure Kid-
ney International Board. In addition, Dr. Kretzler has a patent PCT/
EP2014/073413 “Biomarkers and methods for progression prediction
for chronic kidney disease” licensed. All other authors have no disclo-
sures to report.
References
1. Ng DK, Pierce CB (2021) Kidney disease progression in children
and young adults with pediatric CKD: epidemiologic perspectives
and clinical applications. Semin Nephrol 41:405–415. https:// doi.
org/ 10. 1016/j. semne phrol. 2021. 09. 002
2. Harada R, Hamasaki Y, Okuda Y, Hamada R, Ishikura K (2022)
Epidemiology of pediatric chronic kidney disease/kidney fail-
ure: learning from registries and cohort studies. Pediatr Nephrol
37:1215–1229. https:// doi. org/ 10. 1007/ s00467- 021- 05145-1
3. Cara-Fuentes G, Smoyer WE (2021) Biomarkers in pediatric
glomerulonephritis and nephrotic syndrome. Pediatr Nephrol
36:2659–2673. https:// doi. org/ 10. 1007/ s00467- 020- 04867-y
4. Gipson DS, Massengill SF, Yao L, Nagaraj S, Smoyer WE,
Mahan JD, Wigfall D, Miles P, Powell L, Lin JJ, Trachtman H,
Greenbaum LA (2009) Management of childhood onset nephrotic
syndrome. Pediatrics 124:747–757. https:// doi. org/ 10. 1542/ peds.
2008- 1559
5. Vivarelli M, Gibson K, Sinha A, Boyer O (2023) Childhood
nephrotic syndrome. Lancet 402:809
6. Angeletti A, Bruschi M, Kajana X, La Porta E, Spinelli S, Caridi
G, Lugani F, Verrina EE, Ghiggeri GM (2023) Biologics in ster-
oid resistant nephrotic syndrome in childhood: review and new
hypothesis-driven treatment. Front Immunol 14:1213203. https://
doi. org/ 10. 3389/ fimmu. 2023. 12132 03
7. Eddy S, Mariani LH, Kretzler M (2020) Integrated multi-omics
approaches to improve classification of chronic kidney dis-
ease. Nat Rev Nephrol 16:657–668. https:// doi. org/ 10. 1038/
s41581- 020- 0286-5
8. Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PX,
Barisoni L, Sampson MG, Kopp JB, Lemley KV, Nelson PJ, Lienc-
zewski CC, Adler SG, Appel GB, Cattran DC, Choi MJ, Contreras
G, Dell KM, Fervenza FC, Gibson KL, Greenbaum LA, Hernan-
dez JD, Hewitt SM, Hingorani SR, Hladunewich M, Hogan MC,
Hogan SL, Kaskel FJ, Lieske JC, Meyers KE, Nachman PH, Nast
CC, Neu AM, Reich HN, Sedor JR, Sethna CB, Trachtman H, Tut-
tle KR, Zhdanova O, Zilleruelo GE, Kretzler M (2013) Design of
the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate
primary glomerular nephropathy by a multidisciplinary approach.
Kidney Int 83:749–756. https:// doi. org/ 10. 1038/ ki. 2012. 428
9. Mariani LH, Eddy S, Al Akwaa FM, McCown PJ, Harder JL, Nair
V, Eichinger F, Martini S, Ademola AD, Boima V, Reich HN,
El Saghir J, Godfrey B, Ju W, Tanner EC, Vega-Warner V, Wys
NL, Adler SG, Appel GB, Athavale A, Atkinson MA, Bagnasco
SM, Barisoni L, Brown E, Cattran DC, Coppock GM, Dell KM,
Derebail VK, Fervenza FC, Fornoni A, Gadegbeku CA, Gibson
KL, Greenbaum LA, Hingorani SR, Hladunewich MA, Hodgin
JB, Hogan MC, Holzman LB, Jefferson JA, Kaskel FJ, Kopp JB,
Lafayette RA, Lemley KV, Lieske JC, Lin JJ, Menon R, Meyers
KE, Nachman PH, Nast CC, O’Shaughnessy MM, Otto EA, Reidy
KJ, Sambandam KK, Sedor JR, Sethna CB, Singer P, Srivastava T,
Tran CL, Tuttle KR, Vento SM, Wang CS, Ojo AO, Adu D, Gip-
son DS, Trachtman H, Kretzler M (2023) Precision nephrology
identified tumor necrosis factor activation variability in minimal
change disease and focal segmental glomerulosclerosis. Kidney
Int 103:565–579. https:// doi. org/ 10. 1016/j. kint. 2022. 10. 023
10. Trautmann A, Boyer O, Hodson E, Bagga A, Gipson DS, Samuel
S, Wetzels J, Alhasan K, Banerjee S, Bhimma R, Bonilla-Felix
M, Cano F, Christian M, Hahn D, Kang HG, Nakanishi K, Safouh
H, Trachtman H, Xu H, Cook W, Vivarelli M, Haffner D; Inter-
national Pediatric Nephrology Association (2023) IPNA clinical
practice recommendations for the diagnosis and management of
children with steroid-sensitive nephrotic syndrome. Pediatr Neph-
rol 38:877–919. https:// doi. org/ 10. 1007/ s00467- 022- 05739-3
11. Jayapandian CP, Chen Y, Janowczyk AR, Palmer MB, Cassol
CA, Sekulic M, Hodgin JB, Zee J, Hewitt SM, O’Toole J, Toro
P, Sedor JR, Barisoni L, Madabhushi A, Nephrotic Syndrome
Study Network (NEPTUNE) (2021) Development and evalua-
tion of deep learning-based segmentation of histologic structures
in the kidney cortex with multiple histologic stains. Kidney Int
99:86–101. https:// doi. org/ 10. 1016/j. kint. 2020. 07. 044
12. Zee J, Liu Q, Smith AR, Hodgin JB, Rosenberg A, Gillespie
BW, Holzman LB, Barisoni L, Mariani LH, Nephrotic Syndrome
Study Network (NEPTUNE), NEPTUNE Members (2022) Kidney
biopsy features most predictive of clinical outcomes in the spec-
trum of minimal change disease and focal segmental glomerulo-
sclerosis. J Am Soc Nephrol 33:1411–1426. https:// doi. org/ 10.
1681/ ASN. 20211 01396
13. Debiec H, Dossier C, Letouze E, Gillies CE, Vivarelli M, Putler RK,
Ars E, Jacqz-Aigrain E, Elie V, Colucci M, Debette S, Amouyel P,
Elalaoui SC, Sefiani A, Dubois V, Simon T, Kretzler M, Ballarin J,
Emma F, Sampson MG, Deschenes G, Ronco P (2018) Transethnic,
genome-wide analysis reveals immune-related risk alleles and phe-
notypic correlates in pediatric steroid-sensitive nephrotic syndrome.
Pediatric Nephrology
J Am Soc Nephrol 29:2000–2013. https:// doi. org/ 10. 1681/ ASN.
20171 11185
14. Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeya-
gunawardena A, Thalgahagoda S, Kale A, Gipson D, Srivastava
T, Lin JJ, Chand D, Hunley TE, Brophy PD, Bagga A, Sinha A,
Rheault MN, Ghali J, Nicholls K, Abraham E, Janjua HS, Omoloja
A, Barletta GM, Cai Y, Milford DD, O’Brien C, Awan A, Belostot-
sky V, Smoyer WE, Homstad A, Hall G, Wu G, Nagaraj S, Wigfall
D, Foreman J, Winn MP, Mid-West Pediatric Nephrology Consor-
tium (2015) HLA-DQA1 and PLCG2 are candidate risk loci for
childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc
Nephrol 26:1701–1710. https:// doi. org/ 10. 1681/ ASN. 20140 30247
15. Jia X, Horinouchi T, Hitomi Y, Shono A, Khor SS, Omae Y, Kojima
K, Kawai Y, Nagasaki M, Kaku Y, Okamoto T, Ohwada Y, Ohta K,
Okuda Y, Fujimaru R, Hatae K, Kumagai N, Sawanobori E, Naka-
zato H, Ohtsuka Y, Nakanishi K, Shima Y, Tanaka R, Ashida A,
Kamei K, Ishikura K, Nozu K, Tokunaga K, Iijima K, Research
Consortium on Genetics of Childhood Idiopathic Nephrotic Syn-
drome in Japan (2018) Strong association of the HLA-DR/DQ locus
with childhood steroid-sensitive nephrotic syndrome in the Japanese
population. J Am Soc Nephrol 29:2189–2199. https:// doi. org/ 10.
1681/ ASN. 20170 80859
16. Gipson DS, Troost JP, Spino C, Attalla S, Tarnoff J, Massengill
S, Lafayette R, Vega-Warner V, Adler S, Gipson P, Elliott M,
Kaskel F, Fermin D, Moxey-Mims M, Fine RN, Brown EJ, Reidy
K, Tuttle K, Gibson K, Lemley KV, Greenbaum LA, Atkinson
MA, Hingorani S, Srivastava T, Sethna CB, Meyers K, Tran C,
Dell KM, Wang CS, Yee JL, Sampson MG, Gbadegesin R, Lin
JJ, Brady T, Rheault M, Trachtman H (2022) Comparing kidney
health outcomes in children, adolescents, and adults with focal
segmental glomerulosclerosis. JAMA Netw Open 5:e2228701.
https:// doi. org/ 10. 1001/ jaman etwor kopen. 2022. 28701
17. Gipson DS, Trachtman H, Waldo A, Gibson KL, Eddy S, Dell KM,
Srivastava T, Lemley KV, Greenbaum LA, Hingorani S, Meyers KE,
Kaskel FJ, Reidy KJ, Sethna CB, Tran CL, Wang CS, Tuttle KR, Oh
G, Neu AM, Brown E, Lin JJ, Yee JL, Roth TM, Troost JP, Gillespie
BW, Sampson MG, Kretzler M, Ju W (2020) Urinary epidermal
growth factor as a marker of disease progression in children with
nephrotic syndrome. Kidney Int Rep 5:414–425. https:// doi. org/ 10.
1016/j. ekir. 2019. 11. 018
18. Sethna CB, Meyers KEC, Mariani LH, Psoter KJ, Gadegbeku CA,
Gibson KL, Srivastava T, Kretzler M, Brady TM (2017) Blood pres-
sure and visit-to-visit blood pressure variability among individuals
with primary proteinuric glomerulopathies. Hypertension 70:315–
323. https:// doi. org/ 10. 1161/ HYPER TENSI ONAHA. 117. 09475
19. Shah PP, Brady TM, Meyers KEC, O’Shaughnessy MM, Gibson
KL, Srivastava T, Zee J, Cattran D, Tuttle KR, Gadegbeku C, Glenn
D, Derebail V, Smith A, Wang CS, Gillespie BW, Bitzer M, Sethna
CB (2021) Association of obesity with cardiovascular risk factors
and kidney disease outcomes in primary proteinuric glomerulopa-
thies. Nephron 145:245–255. https:// doi. org/ 10. 1159/ 00051 3869
20. Wang L, Smith-Salzberg B, Meyers KE, Glenn DA, Tuttle KR, Der-
ebail VK, Brady TM, Gibson K, Smith AR, O’Shaughnessy MM,
Srivastava T, Hall G, Zee J, Bitzer M, Sethna CB (2023) Tobacco
exposure in adults and children with proteinuric glomerulopathies:
a NEPTUNE cohort study. BMC Nephrol 24:30. https:// doi. org/ 10.
1186/ s12882- 023- 03073-w
21. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S,
Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ,
Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S,
Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman
NA, Group SS, Hildebrandt F (2015) A single-gene cause in 29.5%
of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol
26:1279–1289.https:// doi. org/ 10. 1681/ ASN. 20140 50489
22. Sampson MG, Gillies CE, Robertson CC, Crawford B, Vega-Warner
V, Otto EA, Kretzler M, Kang HM (2016) Using population genetics
to interrogate the monogenic nephrotic syndrome diagnosis in a case
cohort. J Am Soc Nephrol 27:1970–1983. https:// doi. org/ 10. 1681/
ASN. 20150 50504
23. Robertson CC, Gillies CE, Putler RKB, Ng D, Reidy KJ, Crawford
B, Sampson MG (2017) An investigation of APOL1 risk genotypes
and preterm birth in African American population cohorts. Nephrol
Dial Transplant 32:2051–2058. https:// doi. org/ 10. 1093/ ndt/ gfw317
24. Ng DK, Robertson CC, Woroniecki RP, Limou S, Gillies CE, Reidy
KJ, Winkler CA, Hingorani S, Gibson KL, Hjorten R, Sethna CB,
Kopp JB, Moxey-Mims M, Furth SL, Warady BA, Kretzler M,
Sedor JR, Kaskel FJ, Sampson MG (2017) APOL1-associated glo-
merular disease among African-American children: a collaboration
of the chronic kidney disease in children (CKiD) and nephrotic syn-
drome study network (NEPTUNE) cohorts. Nephrol Dial Transplant
32:983–990. https:// doi. org/ 10. 1093/ ndt/ gfw061
25. Sampson MG, Robertson CC, Martini S, Mariani LH, Lemley KV,
Gillies CE, Otto EA, Kopp JB, Randolph A, Vega-Warner V, Eich-
inger F, Nair V, Gipson DS, Cattran DC, Johnstone DB, O’Toole JF,
Bagnasco SM, Song PX, Barisoni L, Troost JP, Kretzler M, Sedor
JR, Nephrotic Syndrome Study Network (2016) integrative genom-
ics identifies novel associations with APOL1 risk genotypes in black
NEPTUNE subjects. J Am Soc Nephrol 27:814–823. https:// doi. org/
10. 1681/ ASN. 20141 11131
26. Beanlands H, Maione M, Poulton C, Herreshoff E, Hladunewich
MA, Hailperin M, Modes MM, An L, Nunes JW, Trachtman H,
Nachman P, Gipson DS (2017) Learning to live with nephrotic syn-
drome: experiences of adult patients and parents of children with
nephrotic syndrome. Nephrol Dial Transplant 32:i98–i105. https://
doi. org/ 10. 1093/ ndt/ gfw344
27. Kidney Disease: Improving Global Outcomes Glomerular Diseases
Work Group (2021) KDIGO 2021 clinical practice guideline for
the management of glomerular diseases. Kidney Int 100:S1–S276.
https:// doi. org/ 10. 1016/j. kint. 2021. 05. 021
28. Wang CS, Troost JP, Greenbaum LA, Srivastava T, Reidy K, Gibson
K, Trachtman H, Piette JD, Sethna CB, Meyers K, Dell KM, Tran
CL, Vento S, Kallem K, Herreshoff E, Hingorani S, Lemley K, Oh
G, Brown E, Lin JJ, Kaskel F, Gipson DS (2019) Text messaging
for disease monitoring in childhood nephrotic syndrome. Kidney Int
Rep 4:1066–1074. https:// doi. org/ 10. 1016/j. ekir. 2019. 04. 026
29. Thakoordeen-Reddy S, Winkler C, Moodley J, David V, Binns-Roe-
mer E, Ramsuran V, Naicker T (2020) Maternal variants within the
apolipoprotein L1 gene are associated with preeclampsia in a South
African cohort of African ancestry. Eur J Obstet Gynecol Reprod
Biol 246:129–133. https:// doi. org/ 10. 1016/j. ejogrb. 2020. 01. 034
30. Hong X, Rosenberg AZ, Zhang B, Binns-Roemer E, David V, Lv
Y, Hjorten RC, Reidy KJ, Chen TK, Wang G, Ji Y, Simpson CL,
Davis RL, Kopp JB, Wang X, Winkler CA (2021) Joint associations
of maternal-fetal APOL1 genotypes and maternal country of origin
with preeclampsia risk. Am J Kidney Dis 77:e871. https:// doi. org/
10. 1053/j. ajkd. 2020. 10. 020
31. Trachtman H, Desmond H, Williams A, Mariani LH, Eddy S, Ju
W, Barisoni L, Ascani HK, Uhlmann WR, Spino C, Holzman LB,
Sedor JR, Gadegbeku C, Subramanian L, Lienczewski C, Manieri
T, Roberts SJ, Gipson DS, Kretzler M, for the NEPTUNE inves-
tigators (2023) Rationale and design of The Nephrotic Syndrome
Network (NEPTUNE) Match in glomerular diseases: Designing the
right trial for the right patient, today. Kidney international (in press)
Publisher's Note Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations.
Springer Nature or its licensor (e.g. a society or other partner) holds
exclusive rights to this article under a publishing agreement with the
author(s) or other rightsholder(s); author self-archiving of the accepted
manuscript version of this article is solely governed by the terms of
such publishing agreement and applicable law.