![| Treatment algorithm for anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Remission is defined by the absence of manifestations of vasculitis and glomerulonephritis disease activity (Birmingham Vasculitis Activity Score for Wegner granulomatosis [BVAS/WG] of 0). For glomerulonephritis (GN), remission is considered as absence of microscopic hematuria and improved proteinuria and glomerular filtration rate. * Based on peripheral B-cell repopulation plus ANCA reappearance. † In patients with rapidly deteriorating kidney function, corticosteriods are often initiated i.v. as pulse doses of 500 to 1000 mg/d methylprednisone and given for 1 to 3 days before converting to an oral formulation. ‡ Consider re-biopsy in order to guide second-line therapy. AZA, azathioprine; CYC, cyclophosphamide; PLEX, plasma exchange; PR3, proteinase 3; RTX, rituximab.](https://www.researchgate.net/profile/Juan-Mejia-Vilet/publication/337600419/figure/fig2/AS:836778051268609@1576514842328/Treatment-algorithm-for-anti-neutrophil-cytoplasmic-antibody-ANCA-associated_Q320.jpg)

Content uploaded by Juan Manuel Mejia-Vilet
Author content
All content in this area was uploaded by Juan Manuel Mejia-Vilet on Dec 16, 2019
Content may be subject to copyright.
OPEN
Management and treatment of glomerular diseases
(part 2): conclusions from a Kidney Disease:
Improving Global Outcomes (KDIGO) Controversies
Conference
Brad H. Rovin
1
, Dawn J. Caster
2
, Daniel C. Cattran
3
, Keisha L. Gibson
4
, Jonathan J. Hogan
5
,
Marcus J. Moeller
6
, Dario Roccatello
7
, Michael Cheung
8
, David C. Wheeler
9
, Wolfgang C. Winkelmayer
10
and Ju
¨rgen Floege
11
; for Conference Participants
12
1
Division of Nephrology, The Ohio State University, Wexner Medical Center, Columbus, Ohio, USA;
2
Department of Medicine, University of
Louisville School of Medicine, Louisville, Kentucky, USA;
3
Toronto General Research Institute, University Health Network, Toronto, Ontario,
Canada;
4
University of North Carolina Kidney Center at Chapel Hill, Chapel Hill, North Carolina, USA;
5
Division of Nephrology, University
of Pennsylvania, Philadelphia, Pennsylvania, USA;
6
Division of Nephrology and Clinical Immunology, Rheinisch-Westfälische Technische
Hochschule, University of Aachen, Aachen, Germany;
7
CMID (Center of Research of Immunopathology and Rare Diseases), and Division of
Nephrology and Dialysis (ERK-Net member), University of Turin, Italy;
8
KDIGO, Brussels, Belgium;
9
University College London, London, UK;
10
Selzman Institute for Kidney Health, Section of Nephrology, Department of Medicine, Baylor College of Medicine, Houston, Texas, USA;
and
11
Division of Nephrology, Rheinisch-Westfälische Technische Hochschule, University of Aachen, Aachen, Germany
In November 2017, the Kidney Disease: Improving Global
Outcomes (KDIGO) initiative brought a diverse panel of
experts in glomerular diseases together to discuss the 2012
KDIGO glomerulonephritis guideline in the context of new
developments and insights that had occurred over the
years since its publication. During this KDIGO Controversies
Conference on Glomerular Diseases, the group examined
data on disease pathogenesis, biomarkers, and treatments
to identify areas of consensus and areas of controversy.
This report summarizes the discussions on primary
podocytopathies, lupus nephritis, anti-neutrophil
cytoplasmic antibody–associated nephritis, complement-
mediated kidney diseases, and monoclonal gammopathies
of renal significance.
Kidney International (2019) 95, 281–295; https://doi.org/10.1016/
j.kint.2018.11.008
KEYWORDS: anti-neutrophil cytoplasmic antibody–associated vasculitis; C3
glomerulopathy; focal and segmental glomerulosclerosis; KDIGO; lupus
nephritis; membranoproliferative glomerulonephritis; minimal change dis-
ease; monoclonal gammopathies of renal significance
Copyright ª2019, The Author(s). Published by Elsevier Inc. on behalf of the
International Society of Nephrology. This is an open access article under the
CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
The Kidney Disease: Improving Global Outcomes
(KDIGO) initiative published its first ever guideline on
glomerular diseases in 2012.
1
Since then our under-
standing of the pathogenesis of glomerular diseases has
markedly advanced, new diagnostic biomarkers have entered
the clinical arena, and many new therapies have been assessed
in clinical trials. Therefore, a conference consisting of about
100 experts from various disciplines (nephrology, pathology,
rheumatology, pediatrics) and organizations (academia,
pharmaceutical industry) was convened on November 17–19,
2017. The goals were to evaluate the progress that has been
made in the evaluation and management of glomerular dis-
eases, assess continuing gaps in knowledge, and identify the
existing guideline recommendations that should be revisited
in the next update. The attendees were especially encouraged
to outline the most controversial aspects of glomerular
diseases.
This second of 2 reports covers the primary podocyto-
pathies, complement-mediated glomerular diseases, lupus
nephritis (LN), anti-neutrophil cytoplasmic antibody
(ANCA)-associated nephritis, and monoclonal gammopathies
of renal significance. Each disease-specific working group was
asked to consider disease terminology, pathogenesis, bio-
markers, treatment, and recommendations for future studies.
Taken together, these 2 conference summaries will lay the basis
for the guideline updating process that began in August 2018.
MCD AND FSGS
Terminology
The terms “minimal change disease”(MCD) and “focal
segmental glomerulosclerosis”(FSGS) remain relevant.
Although there may be pathophysiologic overlap between
MCD and FSGS, the presence of focal and segmental sclerosis
by light microscopy has diagnostic and prognostic importance.
To discriminate between MCD and FSGS by kidney biopsy, at
Correspondence: Brad H. Rovin, Division of Nephrology, The Ohio State
University, Wexner Medical Center, 395 West 12th Avenue, Ground Floor,
Columbus, Ohio 43210, USA. E-mail: rovin.1@osu.edu, or Jürgen Floege, Di-
vision of Nephrology and Clinical Immunology, Rheinisch-Westfälische
Technische Hochschule, University of Aachen, Pauwelsstrasse 30, 52057
Aachen, Germany. E-mail: jfloege@ukaachen.de
12
See Appendix for list of other Conference Participants.
Received 26 June 2018; revised 30 October 2018; accepted 1 November
2018
www.kidney-international.org KDIGO executive conclusions
Kidney International (2019) 95, 281–295 281
least 20 glomeruli are needed, and biopsies performed soon
after diagnosis may only show MCD, but the patients may later
develop FSGS.
2
However, in children, kidney biopsy is not
usually performed in patients that respond to prednisone
treatment. Response to prednisone treatment and timing of
relapses allows classification of childhood nephrotic syn-
drome.
3
There was consensus that the designations “steroid-
sensitive”and “steroid-resistant”nephrotic syndrome are
clinically useful disease descriptions in children and that most
steroid-sensitive idiopathic nephrotic syndromes in children
are MCD. Response to therapy is often of more prognostic
value than biopsy histology.
The terms “primary/idiopathic FSGS”should be reserved
for FSGS caused by as yet unknown permeability factors.
Patients with genetic, adaptive (in the setting of reduced
nephron mass), drug-induced, and viral-induced FSGS
should not be designated as primary.
4
Primary FSGS is often
characterized by acute-onset heavy proteinuria and diffuse
podocyte foot process effacement histologically. Other FSGS
subtypes typically show more modest proteinuria and
segmental foot process effacement.
5
Further efforts are war-
ranted to better define these FSGS subgroups in the context of
their presumed pathogenesis.
Pathogenesis of MCD and primary/idiopathic FSGS
A role for dysfunctional T cells in MCD was proposed over 40
years ago.
6
More recently, a role for B cells has become
evident, supported by efficacy of immunoadsorption and B-
cell depletion in inducing remission.
3
Thus far none of the reported circulating permeability factor
candidates have been independently validated in primary
FSGS.
7
Soluble urokinase-type plasminogen activator receptor
may be a novel prognostic biomarker for chronic kidney dis-
ease, but does not appear to have a role as a diagnostic
biomarker or to represent the permeability factor in FSGS.
8
Cardiotrophin-like cytokine-1, a member of the inter-
leukin 6 cytokine family, may be a candidate FSGS perme-
ability factor. Cardiotrophin-like cytokine-1 has been
identified in the plasma of patients with FSGS and has been
found to decrease nephrin expression in podocyte culture. In
patients with recurrent FSGS, its concentration may be up to
100 times that of normal subjects. Angiopoietin-like-4, a
secreted glycoprotein, is highly upregulated in the serum and
in podocytes in experimental models of MCD and in the
human disease. This biomarker has relevant potential in pa-
tients with steroid-sensitive nephrotic syndrome.
9,10
It has been suggested that MCD/FSGS may be mediated by
podocyte CD80 (B7-1) expression induced after an innocuous
event such as an infection.
11
However, CD80 overexpression
on podocytes could not be confirmed.
12
A role for glomerular
parietal epithelial cells in the pathogenesis of virtually all
histological types of FSGS lesions has also been proposed.
13
Biomarkers and prediction of prognosis
There are no validated biomarkers ready for clinical use in
MCD or FSGS. The histological subtype of FSGS as defined
by the Columbia classification
14
may support decision
making and help with anticipating response to treatment and
prognosis,
15
but it is not specific for underlying disease
mechanisms. Immunostaining of kidney biopsy specimens for
parietal epithelial cell activation markers may improve
sensitivity for detecting sclerotic lesions when distinguishing
primary FSGS from MCD.
16
Proteomic analysis of kidney
biopsy specimens may provide additional insights.
Genetic testing
Genetic testing for pediatric nephrotic syndrome and adult
FSGS is controversial, but it should be considered for patients
with congenital and infantile forms of nephrotic syndrome
(children <1 year of age) or less than 2 years of age with
steroid-resistant nephrotic syndrome, nephrotic syndrome
associated with other syndromic features, or familial forms of
steroid-resistant nephrotic syndrome/FSGS.
17–19
Adding to
the controversy of when to perform genetic testing, single
gene mutations have been found in up to 30% of patients
under age 25.
18
Testing should target relevant genes based on
patient characteristics and contemporary knowledge. The role
of high-risk apolipoprotein L1 genotypes in the development
of glomerulosclerosis is still under investigation and the
conference attendees agreed that data are still insufficient to
support using this information to guide clinical decisions.
Genetic testing may be considered for inclusion and stratifi-
cation in clinical trials. Biospecimens should routinely be
collected, and patients consented for later genetic analysis.
Ethical issues should be addressed before recommending
genetic analyses.
Treatment
General. While immunomodulatory therapies are the
first-line treatment in primary/idiopathic FSGS caused by a
permeability factor, other FSGS subtypes respond better to
blood pressure control and correction of abnormal glomer-
ular hemodynamics, such as glomerular hypertension (e.g., in
adaptive FSGS), or other specific interventions. The use of
immunomodulatory therapy after causative FSGS mutations
are identified is controversial. Rare reports of varying degrees
of remission in these patients may or may not reflect the
nonimmune modulating effects of these therapies.
20
Following identification of causative mutations, treatment
should include known directed therapies for specific muta-
tions (e.g., coenzyme Q-10, vitamin B12 where applicable),
18
antiproteinuric therapy, and prompt discontinuation of
immunosuppressive therapy in those with no early signal of
response.
21
Pediatric. Because w80% of children with incident
nephrotic syndrome have MCD on biopsy and of the remaining
patients, some will respond to corticosteroid therapy, the con-
ference attendees agreed that there are no data to challenge the
practice of treating all pediatric nephrotic patients with corti-
costeroids first, except those younger than 9 to 10 months of
age. Due to the increased incidence of steroid-resistant
nephrotic syndrome and FSGS with age, consideration to bi-
opsy children older than 12 years prior to treatment is
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
282 Kidney International (2019) 95, 281–295
recommended. In children with steroid-sensitive nephrotic
syndrome, recent data from randomized controlled trials do not
support steroid exposure beyond 8 to 12 weeks.
22–24
Controversies remain about the minimum duration of
corticosteroid therapy required to define steroid resistance.
The 2012 KDIGO guideline recommended at least 8 weeks of
corticosteroids in children before defining steroid resistance.
While consensus was not reached, the need for a globally
accepted definition of “steroid resistance”to improve
comparability of future clinical trials was recognized.
The efficacy of low-dose daily corticosteroids over alternate
day dosing for maintaining remission in relapsing nephrotic
syndrome is promising.
25
Therapy with alternative immu-
nosuppressive agents should be considered in children with
frequently relapsing nephrotic syndrome. While data support
the use of cyclophosphamide (CYC), levamisole, mycophe-
nolate mofetil (MMF), calcineurin inhibitors (CNIs), and
rituximab (RTX), the precise order of therapy is not well
determined.
26
Data are emerging to support an early role of
RTX in the management of children with steroid-dependent
nephrotic syndrome. A direct action of RTX on podocytes
was not confirmed, supporting B-cell depletion as RTX’s
primary mechanism of action.
27
Post hoc analyses suggest that
targeting higher area under the serum/plasma concentration-
time curves for MMF could result in similar numbers of
children maintaining remission with MMF as with CNIs, but
this needs to be confirmed in randomized controlled trials.
28
Nephrotic patients have a high risk of infection regardless
of immunosuppression. The 2012 KDIGO guideline provides
some recommendations regarding vaccinations in children
but does not highlight the importance of hepatitis B screening
and vaccination, especially in those receiving B-cell depleting
therapies.
29
Vaccination against meningococci should also be
included as based on expert opinion.
Adult. In adults, recommending a minimum duration of
16 weeks of high-dose corticosteroids as first-line therapy for
FSGS or MCD was felt to be controversial, given its potential
for toxicity. However, data to support alternative first-line
agents or combination therapies with lower doses of corti-
costeroids are insufficient. The conference attendees agreed
that CNIs or CYC should remain as second-line agents in
adults with frequently relapsing or steroid-dependent MCD.
RTX is an emerging second-line therapy in MCD in adults
although evidence is observational only. The recommenda-
tion for CNIs and MMF as second- and third-line treatments,
respectively, for FSGS should be maintained. Randomized
controlled trials are underway to investigate the value of RTX
in adult MCD (Efficacy of Rituximab in Comparison to
Continued Corticosteroid Treatment in Idiopathic Nephrotic
Syndrome; NCT03298698) and the CD80 inhibitor abatacept,
regardless of CD80 expression on podocytes, in MCD and
FSGS (Pilot Study to Evaluate the Safety and Efficacy of
Abatacept in Adults and Children 6 Years and Older With
Excessive Loss of Protein in the Urine Due to Either Focal
Segmental Glomerulosclerosis or Minimal Change Disease;
NCT02592798).
Future studies
It will be important to distinguish between primary and
secondary FSGS for clinical trials and include only those
patients for which an investigational therapy may be effective.
Immunologic therapies should focus on primary FSGS;
antifibrotic therapies could recruit all forms of FSGS. Rec-
ommendations from the 2012 guideline that should be
revisited are outlined in Supplementary Table S1.
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
Terminology and diagnosis
While the term “membranoproliferative”glomerulonephritis
(GN) retains value as a histologic descriptor of glomerular
injury, our increasing understanding of C3 glomerulopathy
(C3G) and the monoclonal gammopathies of renal signifi-
cance (MGRS) (paraprotein-associated kidney diseases)
illustrate the need for nomenclature based on pathogenesis
and injury pattern. Additionally, the histology of these specific
etiologies is not always membranoproliferative GN. There-
fore, updated clinical practice guidelines should emphasize a
diagnostic approach to GN that considers both pathobiology
and renal histology, such as that outlined in Table 1.
30
Over
time, and with greater understanding of these diseases, using
such a scheme may lead to the elimination of mem-
branoproliferative GN as a distinct category of GN in clinical
practice guidelines. The following discussion highlights con-
troversies that exist within this framework, including the issue
of overlapping disease mechanisms, the conundrum of
common kidney biopsy features, and the inevitable fact that
some cases will remain “idiopathic”in nature.
31
The use of nonroutine histological techniques, including
pronase to unmask hidden epitopes,
32–35
C4d staining to
distinguish C3G from Ig-mediated and postinfectious GN,
36
and staining for the DNA J homolog subfamily B member
9 protein in fibrillary GN
37,38
may also help in the diagnosis
and possibly in understanding the pathogenesis of GN with
an membranoproliferative glomerulonephritis pattern. Most
of these techniques need additional verification.
C3 Glomerulopathies
Pathogenesis. C3G is caused by abnormal complement
activation, deposition and/or degradation. Drivers of disease
are reviewed in recent consensus
39
and KDIGO Controversies
Conference reports.
40
While it was generally agreed that human and animal data
support the role of complement in well-described scenarios,
as a matter of practicality it continues to be difficult to sub-
stantiate the causal role of either single nucleotide changes or
C3 nephritic factors in the majority of cases.
41,42
Further-
more, the interpretation of published data is confounded by
heterogeneity in the kidney biopsy criteria used for diagnosis.
Addressing these controversies is especially important given
that targeted anticomplement agents are available.
Biomarkers and prediction of prognosis. The role of bio-
markers in the diagnosis and management of C3G has been
summarized recently.
40
The utility of biomarkers such as
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 283
soluble C5b-9 levels for predicting treatment response re-
mains unclear. Controversy remains regarding the clinical
utility of an extended biomarker assessment at diagnosis, and
the use of serial complement testing requires further study.
Testing for paraproteins in C3G has also received increased
attention.
43
Treatment. A contemporary approach to the treatment of
C3G has been outlined,
40
derived mostly from case reports
and retrospective case series. An important knowledge gap in
the treatment of C3G is the absence of a robust understanding
of its natural history. Current treatments have been empiri-
cally extrapolated from other glomerular diseases. The
optimal duration of therapy remains unclear. Current treat-
ment guidelines focus on inhibiting definable pathways
(inflammation or terminal complement activity) with avail-
able targeted therapeutics (antiproliferative agents or terminal
complement blockers). Treatment of active disease with MMF
and corticosteroids has shown promise in 2 retrospective case
series,
44,45
but was not found to be effective in a third case
series in patients with more severe baseline kidney disease.
46
For patients with C3G and monoclonal gammopathy, a
recent retrospective case series found superior hematologic
and renal response rates, as well as renal survival, for patients
treated with clone-directed chemotherapy compared with
conservative or immunosuppressive treatment.
47
Monoclonal Gammopathies of Renal Significance
Pathogenesis. Preclinical and clinical studies have eluci-
dated the pathogenesis of some paraprotein-associated kidney
diseases. For example, heavy chain deposition disease is
caused by a truncated Ig heavy chain that lacks the first
constant domain (CH1 deletion).
48,49
Specific physiochemical
properties of the truncated heavy chain may explain its
tropism for the kidney.
50
Most patients with heavy chain
deposition disease have an underlying plasma cell clone that
does not meet criteria for multiple myeloma (i.e., a MGRS),
and evidence of the truncated heavy chain can be found in the
serum and bone marrow.
50
In MGRS, pathogenic Igs are from plasma cell or B-cell
clones. Targeting these clones may improve outcomes,
47,50,51
but the clones are often undetectable. The International Kid-
ney and Monoclonal Gammopathy Research Group recom-
mends that all patients with paraprotein-associated kidney
disease undergo hematology evaluation, including a bone
marrow biopsy, but the utility of the bone marrow is not clear
in patients without a detectable circulating paraprotein.
47,52–54
Biomarkers and prediction of prognosis. In multiple
myeloma and light chain amyloidosis, achieving hematologic
response (improvement in levels of circulating paraprotein) is
associated with improved overall and renal survival.
55–58
Moreover, stabilization or improvement in kidney function
and proteinuria may be linked with long-term renal survival.
59
There are emerging data regarding the importance of hema-
tologic response in MGRS,
50,51,60
but it is not clear how to
monitor patients without a detectable circulating paraprotein
beyond glomerular filtration rate (GFR) and proteinuria.
Treatment. The International Kidney and Monoclonal
Gammopathy Research Group published an approach to man-
aging MGRS based on expert opinion.
61
Risk stratification was
based on kidney dysfunction and proteinuria, and treatment
strategies utilized a clone-directed approach similar to that
employed for multiple myeloma and lymphomas (i.e., chemo-
therapeutic regimens, autologous stem cell transplant). A large
retrospective case series found that using bortezomib-based
Table 1 | A pathogenesis-based approach to glomerulonephritis
Pathogenic type Disease examples
Immune complex glomerulonephritis IgA nephropathy
Lupus nephritis
Fibrillary glomerulonephritis (polyclonal/DNAJB9-positive subtype)
Infection-associated glomerulonephritis
Mixed (types II and III) cryoglobulinemic glomerulonephritis
Pauci-immune glomerulonephritis ANCA-associated vasculitis
ANCA-negative pauci-immune glomerulonephritis
Antiglomerular basement membrane glomerulonephritis Antiglomerular basement membrane disease
Monoclonal Ig-associated glomerulonephritis Monoclonal Ig deposition disease (LCDD, HCDD, LHCDD)
Proliferative glomerulonephritis with monoclonal Ig deposits
Monoclonal (type I) cryoglobulinemic glomerulonephritis
Immunotactoid glomerulopathy
Fibrillary glomerulonephritis (monoclonal subtype)
Complement-mediated glomerulonephritis C3 glomerulonephritis
Dense deposit disease
ANCA, anti-neutrophil cytoplasmic antibody; DNAJB9, DNA J homolog subfamily B member 9; HCDD, heavy chain deposition disease; LCDD, light chain deposition disease;
LHCDD, light and heavy chain deposition disease.
Adapted from Sethi S, Haas M, Markowitz GS, et al. Mayo Clinic/Renal Pathology Society Consensus report on pathologic classification, diagnosis, and reporting of GN. J Am Soc
Nephrol. 2016;27:1278–1287,
30
with permission. Copyright ª2016 the American Society of Nephrology.
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
284 Kidney International (2019) 95, 281–295
therapy for monoclonal Ig deposition disease led to higher he-
matologic and renal response rates, and prolonged renal survival,
compared with results from previously published literature.
51
As
noted previously, clone-directed chemotherapy results in
improved hematologic and renal outcomes for patients with
paraprotein-associated C3G compared with other immunosup-
pression or conservative treatment.
47
Controversy exists regarding treatment of patients without
a detectable underlying clone, but recent uncontrolled data
suggest benefit from empiric chemotherapy.
53
A multidisci-
plinary, onco-nephrologic approach to patients with MGRS is
recommended.
62
Hepatitis C-associated glomerulonephritis. The KDIGO
Clinical Practice Guideline on the Prevention, Diagnosis, Eval-
uation and Treatment of Hepatitis C in CKD summarizes an
approach to the treatment of these patients
63
(Table 2). This
approach will require validation. The development or
persistence of cryoglobulinemic vasculitis (with or without
kidney involvement) after achieving sustained virologic
response has been described.
64–67
Whether this presentation
reflects continued B-cell production of pathogenic immune
complexes requires further study.
Fibrillary GN. Immunohistochemical staining on kidney
biopsy for the DNA J homolog subfamily B member 9 protein
was identified as a sensitive and specific marker in fibrillary
GN.
37,38
The role of DNA J homolog subfamily B member 9
in disease pathogenesis is unknown. The data on treating
fibrillary GN consist of small studies using a variety of ther-
apies, none of which have been conclusive.
68–72
Future studies. Investigations considered critical to the
development of management protocols for C3G, immune-
complex GN, and monoclonal gammopathies of renal sig-
nificance are outlined in Table 3. Recommendations from the
2012 guideline that should be revisited are outlined in
Supplementary Table S2.
LUPUS NEPHRITIS
Terminology
LN is histologically classified by the International Society of
Nephrology/Renal Pathology Society system,
73
but this clas-
sification does not consider tubulointerstitial injury, vascular
lesions, or podocytopathies.
74–76
Patients with tubulointer-
stitial injury, thrombotic microangiopathy (TMA), and renal
vasculitis have worse outcomes.
74,75,77–80
Additionally, the
International Society of Nephrology/Renal Pathology Society
classification lacks sufficient quantification of disease activity
and chronicity, and descriptive categories lack clear prog-
nostic value. An evidence-based approach is needed to better
define clinically relevant categories within the class III/IV
spectrum, including the significance of segmental necrotizing
lesions,
81,82
along with the development of LN activity and
chronicity indices that accurately identify patients who would
benefit from immunosuppression. An international working
group of leading nephropathologists recently proposed up-
dates to the International Society of Nephrology/Renal Pa-
thology Society classification system to address limitations
within the current system.
83
According to the Systemic Lupus International Collabo-
rating Clinic diagnostic criteria for systemic lupus erythema-
tosus (SLE), immune complex GN consistent with LN in the
setting of a positive antinuclear antibody or anti–double-
stranded DNA is sufficient for diagnosing SLE.
84
Systemic
Lupus International Collaborating Clinic criteria demonstrated
increased sensitivity when compared with American College of
Rheumatology classification with similar specificity in the
validation cohort.
84
However, when applied to a cohort of
patients with immune complex GN, the Systemic Lupus In-
ternational Collaborating Clinic criteria demonstrated
decreased specificity compared with those of the American
College of Rheumatology, with some patients incorrectly
identified as having SLE.
85
Nonetheless, the Systemic Lupus
International Collaborating Clinic criteria allow giving patients
with lupus-like conditions a label, which may help in coping
with disease and for insurance and medication coverage.
Pathogenesis
The pathogenesis of LN involves genetic, epigenetic, immu-
noregulatory, hormonal, and environmental phenomena.
86
Multiple gene polymorphisms have been associated with an
increased risk of SLE and/or LN;
87
many of them involve im-
mune cells and immunoregulatory pathways.
86–88
Presently,
there is no clear clinical benefit from genetic testing. However,
identification of these polymorphisms has given insight into
pathways involved in the pathogenesis of LN.
87,89,90
LN patients
of African ancestry with apolipoprotein L1 risk alleles are at
increased risk for worse renal outcomes;
91
however, APOL1
Table 2 | KDIGO clinical practice guideline on the treatment of HCV-associated glomerulonephritis
Renal presentation Treatment
Stable kidney function and/or nonnephrotic proteinuria Direct-acting antiviral therapy
Cryoglobulinemic flare, nephrotic syndrome,
or rapidly progressive kidney failure
Direct-acting antiviral therapy with immunosuppressive treatment, with
or without plasma exchange
Histologically active HCV-associated glomerulonephritis that
does not respond to direct-acting antiviral therapy
Rituximab as first-line immunosuppressive treatment
HCV, hepatitis C virus; KDIGO, Kidney Disease: Improving Global Outcomes.
From the KDIGO Hepatitis C Work Group.
63
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 285
testing is not routinely available and the risks and benefits of
APOL1 testing need to be clarified.
Biomarkers and prediction of prognosis
Proteinuria, hematuria, urinary sediment, and estimated
GFR. No single biomarker predicts the development of LN
in patients with SLE or of LN flares in patients with quiescent
disease. Proteinuria, hematuria, urinary sediment analysis,
and serum creatinine (with estimated GFR)
92
remain
important to diagnose LN and monitor response to therapy.
The diagnosis of LN should be confirmed by biopsy.
There are limitations to these clinical markers. Repeat
kidney biopsy studies have shown that patients with resolu-
tion of proteinuria and normalization of serum creatinine can
still have histologic activity on biopsy and vice versa.
93–97
Studies are needed to evaluate the clinical relevance of this
discordance.
Proteinuria at 1 year was the best predictor of long-term
renal outcome.
98–100
Random spot urine protein-to-
creatinine ratios are not sufficiently accurate to direct thera-
peutic changes. Such changes should be based on 24-hour
urine collections for proteinuria or the urine protein-to-
creatinine ratios from a 24-hour urine.
101
Anti–double-stranded DNA, complement C3, C4, anti-C1q tes-
ting. The combination of elevated anti–double-stranded
DNA, low serum complement, and anti-C1q autoantibody
levels, if available, is strongly associated with renal involve-
ment in SLE and should be monitored in patients at risk for
LN or LN flare.
102,103
Levels may change several months prior
to LN flare, and how these changes relate to flare prediction
needs to be validated in prospective studies.
Novel urine/serum biomarkers. Several putative novel
serum and urine biomarkers have been studied in LN.
104–107
These candidate markers must be studied in a prospective
fashion, ideally in clinical trials. It is likely that biomarker
panels will be required to accurately stratify risk, predict flare,
determine treatment, monitor response to treatment, and
predict prognosis. Molecular interrogation of the kidney bi-
opsy may help in these processes.
108–110
Treatment
Antimalarials. Antimalarial treatment is recommended for
all patients with LN. Observational and cohort studies have
demonstrated that antimalarials reduce the odds of developing
LN in patients with SLE and are associated with a higher like-
lihood of a complete renal response to treatment and a reduced
likelihood of developing end-stage kidney disease.
111–114
Corticosteroids. Corticosteroids, although almost universal
in LN regimens, are associated with significant short- and long-
term adverse effects. Patients with LN are more likely to develop
corticosteroid-associated organ damage than are SLE patients
without nephritis.
115
Moderate doses are not safer and are
associated with as many adverse effects as high doses are.
116
Therefore, although not possible for all patients, an attempt
to minimize corticosteroids (e.g., prednisone equivalent #5
mg/d) during LN maintenance therapy, should be made. Reg-
imens with reduced or no oral corticosteroids and rapid
tapering protocols are under investigation
93,117,118
(Aurinia
Renal Response in Active Lupus With Voclosporin [AURORA],
NCT03021499; Safety and Efficacy of Two Doses of Ani-
frolumab Compared to Placebo in Adult Subjects With Active
Proliferative Lupus Nephritis [TULIP-LN1], NCT02547922).
Immunosuppressive therapy. While CYC- or MMF-based
regimens for remission induction remain the gold standard
therapy for most patients, CNI-based regimens have been
studied in Asia, and they often combine MMF and corticoste-
roids with a CNI.
119
A large Chinese multicenter RCT compared
low-dose MMF, tacrolimus, and corticosteroids with monthly
i.v. CYC and corticosteroids for induction therapy of LN. The
CNI-based regimen was superior at achieving 24-week complete
and partial renal remissions.
119
However, the cumulative
response rates were similar in the 2 treatment arms with
extended follow-up.
120
Ongoing studies are addressing the role
and toxicity of CNI-based regimens in ethnically diverse pop-
ulations. Protocol biopsies in clinical trials using CNIs will help
clarify immunologic responses as CNIs can reduce proteinuria
by nonimmunologic mechanisms.
Maintenance treatment. Maintenance treatment after in-
duction typically consists of MMF or azathioprine (AZA)
with or without low-dose corticosteroids. It is not clear how
long to continue maintenance. In recent clinical trials, the
duration of maintenance has been 3 to 5 years, and many
patients remained on maintenance therapy for 10 years.
121,122
A minimum of 3 years of maintenance is suggested. A
maintenance withdrawal trial is underway (Randomized
MMF Withdrawal in Systemic Lupus Erythematosus [ALE06];
NCT01946880). Prolonged maintenance for “high-risk”
groups (Table 4) may be considered.
Preliminary studies suggest that intensive B-cell depletion
with a RTX plus CYC-based regimen may avoid the need for
maintenance therapy.
117
This must be verified in large studies.
Table 3 | Examples of future directions in studying C3
glomerulopathy, immune complex glomerulonephritis, and
monoclonal gammopathies of renal significance
C3 glomerulopathy
Establish trends in complement abnormalities with repeated testing
and in the setting of treatment
Evaluate the association of baseline kidney biopsy findings with clinical
outcomes and subsequent changes in kidney biopsy histology
Explore the association of complement testing abnormalities with
response to specific anticomplement therapies
Immune complex glomerulonephritis
Determine the diagnostic, prognostic and therapeutic value of com-
plement testing
Determine whether complement abnormalities are pathogenic or
reflective of disease activity
Identify glomerular antigens involved in pathogenesis
Monoclonal gammopathies of renal significance
Define the value of performing bone marrow biopsies in patients
without detectable paraproteinemia
Perform randomized controlled trials comparing the efficacy and safety
of clone-directed therapies
Elucidate the role of autologous stem cell transplantation
Determine optimal treatment for patients without detectable clones
Ascertain the role for maintenance therapy
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
286 Kidney International (2019) 95, 281–295
Slowly withdrawing immunosuppression could be
considered in patients with complete clinical remission. A
repeat kidney biopsy may be helpful to exclude persistent but
clinically silent histologic activity. Patients should be closely
monitored for relapse after decreasing or discontinuing
maintenance therapy.
123–127
Refractory disease. LN may be considered refractory if a
patient does not respond to either of the currently standard
induction therapies (CYC or MMF) used sequentially. A
suggested algorithm for refractory disease is illustrated in
Figure 1. Medication adherence should always be evaluated.
Repeat kidney biopsy to distinguish active LN from scarring
and/or identify new lesions could be considered. For persis-
tently active LN, if MMF was used for induction, consider
switching to CYC or vice versa. After this, RTX or CNI-based
regimens could be tried.
117,119,128–131
Special circumstances
Class V LN. There is consensus that class V LN with persis-
tent nephrotic proteinuria should receive immunosuppression,
but some would also treat patients with lower levels of protein-
uria.
132
The level of proteinuria at which immunosuppressive
therapy may provide benefit therefore needs to be established.
Class V LN is often treated initially with MMF, but if not effective,
CYC may be used. Some investigators also suggest using CNIs for
class V LN. RTX may be considered in the treatment options for
class V LN.
133
TMA. TMA with LN on kidney biopsy may be due to
antiphospholipid antibodies/syndrome ([APS], anti-
cardiolipin antibodies, anti-
b
2 glycoprotein I, and lupus
anticoagulant), thrombotic thrombocytopenic purpura, or
atypical hemolytic uremic syndrome.
134,135
Treatment should
be guided by the underlying etiology of TMA.
134
Plasma ex-
change is indicated for thrombocytopenic purpura, but it may
also be beneficial in cases of refractory APS.
136,137
Anti-
complement therapies may be considered in catastrophic
APS, thrombocytopenic purpura, complement-mediated
TMA, and recurrent TMA in an allograft.
138–141
Anti-
coagulation remains the standard of care when APS is pre-
sent.
142
However, the impact of anticoagulation on renal
1
• Verify adherence (check mycophenolic level if on MMF/check infusion
records if on CYC)
2
• Repeat biopsy if concern for chronicity or other diagnosis (e.g., TMA)
3
• Switch from MMF to CYC or vice versa
4
• Consider regimen with combined MMF/CNI “multi-target” therapy or
• Addition of rituximab or
• Consider prolonged course of i.v. pulse CYC
5
• Consider i.v. Ig or plasmapheresis (especially in setting of
concomitant TMA or refractory APS) though there is minimal evidence
outside of case reports
Figure 1 | Algorithm for refractory disease in lupus nephritis. APS, antiphospholipid syndrome; CNI, calcineurin inhibitor; CYC,
cyclophosphamide; MMF, mycophenolate mofetil; TMA, thrombotic microangiopathy.
Table 4 | | Lupus nephritis patients at high risk for poor renal outcome (risk increases with the number of risk factors present)
Patient characteristics Serologic characteristics Histologic characteristics
African or Hispanic ancestry
Male
Pediatric onset
Frequent relapses
Incomplete remission
Neuropsychiatric lupus
Proteinuria >4 g/d at diagnosis
Antiphospholipid antibodies
or antiphospholipid syndrome
Persistent hypocomplementemia
High titer dsDNA antibodies
High titer C1q antibodies
Crescentic glomerulonephritis
Thrombotic microangiopathy
Extensive tubulointerstitial damage
dsDNA, double-stranded DNA.
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 287
lesions is unclear, and many patients experience a decline in
kidney function despite therapeutic anticoagulation.
143
Mammalian target of rapamycin inhibition increased kidney
transplant survival in patients with a history of APS ne-
phropathy, but further studies are needed in native kidneys.
144
Pregnancy. Patients who are on MMF maintenance and
wish to become pregnant should be switched to AZA, as
MMF is teratogenic. Similarly, renin-angiotensin system
blockers should be stopped before conception. CNIs may be
considered for treatment of LN in pregnancy if AZA cannot
be tolerated, as adjunct therapy with AZA in severe cases or as
primary therapy for class V LN with nephrotic syndrome.
145
Posttransplant. Patients with LN have equivalent or better
outcomes following kidney transplantation compared with other
primary glomerular diseases.
146
Clinically significant LN post-
transplant recurs in <20% of patients.
147–151
Patients should
remain on hydroxychloroquine posttransplant and be on MMF/
CNI-based immunosuppressive regimen. Patients with mild
flares can be treated with oral corticosteroids alone. Patients with
moderate flares should be treated with i.v. corticosteroids and
increased MMF. Patients with crescentic disease/severe flare
should be treated with i.v. corticosteroids and CYC. MMF
should be held while patient is on CYC therapy.
Pediatric-onset disease. Pediatric-onset LN, occurring
before age 16, needs further study but children are excluded from
adultLNtrials.Childrenoftenhavefewcomorbidities,butthey
exhibit more severe disease with a higher genetic contribution.
There is consensus for response, relapse, and treatment for chil-
dren with proliferative LN.
152
Children with class V LN tend to
need additional immunosuppression even with subnephrotic
proteinuria.
153,154
The Single Hub and Access Point for Paediatric
Rheumatology in Europe initiative has recently published rec-
ommendations for the treatment of children with LN.
155
Future studies
Class III/IV LN should be studied separately from class V LN
in view of their different disease courses. Data are needed to
assess benefits of treating class V LN patients with sub-
nephrotic proteinuria. Validated histological activity indices
are also needed. Clinical trials should require a recent (#3
months) kidney biopsy, and trial duration should be at least
12 months for induction therapies and longer to assess relapse
rates. Patient-reported outcomes should be integrated into
future studies and biomarkers of prognosis and response are
needed. Recommendations from the 2012 guideline that
should be revisited are outlined in Supplementary Table S3.
ANCA-ASSOCIATED VASCULITIS
Terminology
ANCA-associated vasculitis (AAV) represents a group of small
vessel vasculitides that include granulomatosis with poly-
angiitis (GPA), microscopic polyangiitis, and eosinophilic
granulomatosis with polyangiitis.
156
Renal-limited vasculitis
can also occur. Kidney histology shows pauci-immune, focal
necrotizing, and crescentic GN. Pauci-immune refers to the
paucity but not absence of immune and complement deposits.
AAV is characterized by ANCA specific for myeloperox-
idase (MPO-ANCA) or proteinase 3 (PR3-ANCA). Rare pa-
tients with pauci-immune GN are negative for ANCA, but
they are considered in the same spectrum of diseases. There is
some evidence that a percentage of these cases may vary in
ANCA detectability when tested by different assays.
157
Classifying patients as having GPA or microscopic polyangiitis
might some provide prognostic information, but ANCA serology
(MPO- or PR3-ANCA) is more relevant as it seems to predict
outcomes and risk of relapse better.
158,159
A genetic component
exists in AAV and genetic distinctions between GPA and micro-
scopic polyangiitis are associated with ANCA specificity.
160
Pathogenesis
The pathogenesis of AAV involves genetic, epigenetic, immu-
noregulatory, hormonal, and environmental phenomena. The
relative contribution of each of these factors may vary in an
individual patient. Polymorphisms associated with an increased
risk of AAV particularly involve the human leukocyte antigen
system (immune regulation) and target antigen (in anti-PR3
disease).
160
A role for complement activation in the pathogen-
esis of ANCA-associated nephritis (AAN) has emerged from
therapeutic studies with complement inhibitors.
161,162
Biomarkers and prediction of prognosis
Proteinuria, hematuria, urinary sediment, and estimated
GFR. Proteinuria, hematuria, urinalysis, estimated GFR, and
kidney biopsy are important clinical tools for the diagnosis and
management of AAN.
163
At present, there is no biomarker that
can be used to predict the development of AAN or disease flares.
ANCA. Both an increase in ANCA titer and persistently
positive ANCA are modestly but significantly associated with
disease relapse, although serial ANCA testing is not suffi-
ciently robust to trigger changes in therapy.
164
Disease
relapse is more frequent in PR3-ANCA than in those
who are MPO-ANCA, and relapse may be predicted by
PR3-ANCA levels.
158,159
Novel urine/serum biomarkers. The Birmingham Vascu-
litis Activity Score and the Vasculitis Damage Index utilize
traditional clinical and laboratory biomarkers to evaluate
vasculitis activity and are valuable research tools.
165,166
However, traditional laboratory measures do not differen-
tiate between active disease and chronic damage very well. In
apost hoc analysis of the Rituximab for ANCA-Associated
Vasculitis (RAVE) study, chemokine C-X-C motif chemo-
kine ligand 13, matrix metalloproteinase-3, and tissue in-
hibitor of metalloproteinases-1 discriminated active from
inactive AAV better than erythrocyte sedimentation rate
and C-reactive protein did.
167
Tissue inhibitor of
metalloproteinases-1 was the best marker of AAV activity as
reported in the Remission Induction Therapy in Japanese
Patients With AAV and Rapidly Progressive Glomerulone-
phritis (RemIT-JAV-RPGN) study.
168
Urinary soluble
CD163 levels are also promising for identifying active renal
vasculitis.
169
These biomarkers need independent and,
ideally, prospective validation.
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
288 Kidney International (2019) 95, 281–295
Treatment
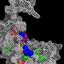
An algorithm for the treatment of AAV is given in Figure 2.
Corticosteroids. Corticosteroids are used almost univer-
sally for AAV and are often given as 500- to 1000-mg i.v.
pulses daily for 1 to 3 days at the initiation of treatment,
especially in patients with a clinical picture of rapidly pro-
gressive GN. However, corticosteroid monotherapy is not
effective and corticosteroids are associated with significant
short- and long-term adverse effects. However complement
inhibition is on the horizon as an adjunct/steroid-sparing
therapy in AAV/AAN.
170
Induction. CYC has been the immunosuppressant of
choice for decades. Despite its efficacy in the management of
AAV, its safety profile has required the need for testing the
value of alternative options. Recently, RTX has been proven to
be as effective as CYC induction/AZA maintenance for AAN
patients with serum creatinine <4 mg/dl (354
m
mol/l).
171–173
An alternative approach includes the use of CYC for the in-
duction phase and considers RTX for maintenance. It is
unknown whether treatment should be different for MPO-
ANCA and PR3-ANCA, however a post hoc analysis of
RAVE suggested RTX was superior to CYC for PR3-ANCA
and as effective as CYC for MPO-ANCA.
174
In a pooled
analysis of the Comparison of Methotrexate or Azathioprine
as Maintenance Therapy for ANCA-Associated Vasculitides
(WEGENT) and RAVE trials, clinical differences between
For new diagnosis, biopsy to investigate
the extent of kidney involvement
CYC with
corticosteroids
RTX with
corticosteroids
Maintenance
AZA for at least
18 months
Remission
Induction
oNseY
New diagnosis or relapse of
ANCA-associated vasculitis
Taper after
24–48 months
RTX
on demand*
RTX on a fixed
schedule for at
least 18 months
Refractory disease:
• No improvement in 4 weeks
• Improvement of less than 50% in 6 weeks
of treatment (as measured by BVAS/WG)
• Chronic persistent disease after more than
12 weeks
Change in therapy‡
‡
:
• Switch to RTX if previously treated with CYC
(especially in PR3–ANCA patients) or vice versa
• Oral CYC if previous i.v. CYC failure
(and RTX unavailable)
• i.v. Ig 0.4 g/kg for 5 days especially if persistent
low disease activity
> 4 mg/dl (354 µmol/l) serum
creatinine or crescentic GN
+ Pulmonary
hemorrhage
CYC with
corticosteroids†
CYC + RTX with
corticosteroids†
PLEX
Rapidly progressive
ANCA-associated vasculitis
+/–
Figure 2 | Treatment algorithm for anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis. Remission is defined by the
absence of manifestations of vasculitis and glomerulonephritis disease activity (Birmingham Vasculitis Activity Score for Wegner granulomatosis
[BVAS/WG] of 0). For glomerulonephritis (GN), remission is considered as absence of microscopic hematuria and improved proteinuria and
glomerular filtration rate.
*
Based on peripheral B-cell repopulation plus ANCA reappearance.
†
In patients with rapidly deteriorating kidney
function, corticosteriods are often initiated i.v. as pulse doses of 500 to 1000 mg/d methylprednisone and given for 1 to 3 days before
converting to an oral formulation.
‡
Consider re-biopsy in order to guide second-line therapy. AZA, azathioprine; CYC, cyclophosphamide; PLEX,
plasma exchange; PR3, proteinase 3; RTX, rituximab.
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 289
MPO- and PR3-ANCA-positive patients with GPA were not
obvious. The risk of relapse was associated more closely with
disease type than ANCA subset.
175
In patients with median GFR <20 ml/min per 1.73 m
2
,a
RTX-based regimen (An International, Randomized, Open
Label Trial Comparing a Rituximab-based Regimen With a
Standard Cyclophosphamide/Azathioprine-based Regimen in
the Treatment of Active, Generalized ANCA-Associated
Vasculitis [RITUXVAS] trial) consisting of a combination of
corticosteroids, RTX 375 mg/m
2
per week for 4 weeks, and 2
i.v. CYC pulses followed by low-dose corticosteroids was
found to be equal to the administration of standard cortico-
steroids with i.v. CYC for 3 to 6 months followed by AZA.
176
At 24 months, the composite outcome of death, end-stage
kidney disease, and relapse did not differ between groups.
Relapses occurred in 21% of patients in the RTX group and
18% of the control group.
177
Maintenance treatment. In AAN, maintenance therapy is
initiated after remission is achieved, usually within 3 to 6
months after beginning induction and typically consists of
AZA or RTX. There is no consensus regarding the length of
maintenance therapy in AAV. Duration may be different
depending on the underlying ANCA serology as well as
treatment, but this has not been adequately studied.
For conventional therapy with CYC induction and AZA
maintenance, the relapse rate was lower if maintenance was
continued for 48 as opposed to 24 months.
178
Alternatively,
patients with MPO-ANCA who achieve remission and ANCA
negativity at end of induction might require a shorter course of
maintenance. This is based on the observation that most pa-
tients with MPO–microscopic polyangiitis given a single
course of 6 rituximab infusions without any maintenance
therapy did not relapse for a mean of 66 months.
179
However,
it is unlikely that this observation applies to MPO-GPA.
175
Retrospective and prospective studies have used RTX for
remission maintenance in AAV, but there has been no
consensus on dosing for maintenance or even induction
therapy (Table 5).
172,177,180-186
It is also not clear whether
rituximab should be given as a fixed regimen or only when B
cells reappear, but this is being tested by the Comparison Study
of Two Rituximab Regimens in the Remission of ANCA-
Associated Vasculitis (MAINRITSAN 2; NCT01731561).
Refractory disease. In a patient with worsening creatinine
and/or proteinuria after initial therapy, medication adherence
should be evaluated. Additionally, a repeat kidney biopsy to
distinguish active AAV from scarring and/or identify new
lesions could be considered. For continued active AAV le-
sions, initial treatment should change to the other standard-
of-care regimen (i.e., switch from CYC to RTX or vice versa).
Special circumstances
Role of plasma exchange. Plasma exchange should be
considered in AAN with severe renal impairment (serum
creatinine >5.6 mg/dl [495
m
mol/l]) and/or diffuse crescents.
Plasma exchange may also have a role in AAV with pulmonary
hemorrhage. The role of plasma exchange in patients with
pulmonary hemorrhage and/or less severe renal impairment is
being studied in the Plasma Exchange and Glucocorticoids for
Treatment of Antineutrophil Cytoplasm Antibody–Associated
Vasculitis (PEXIVAS) trial (NCT00987389).
Childhood-onset disease. AAV in children should be stud-
ied separately from AAV in adults.
187
Pediatric scoring tools for
disease activity and damage have been developed. There is a
high frequency of kidney disease (75%) among pediatric AAV
patients and a predominance of female subjects (65%,
compared with 40%–45% in adult cohorts).
188
There have not
been any randomized controlled trials in children, but cohort
studies support efficacy of both CYC and RTX.
189,190
Future studies
Future studies should further investigate the CYC-sparing
effect of biological agents (e.g., anti-B-cell therapies) and
the steroid-sparing effect of newer agents, such as comple-
ment inhibitors.
The clinical role and the cost-effectiveness of RTX in severe
kidney disease and optimal maintenance regimens remain
undefined.
Future clinical trials in AAV should target subgroups of
patients stratified according to ANCA subtype, identification
of high-risk patients (e.g., with comorbidities), and differ-
entiation between active versus chronic disease by noninvasive
biomarkers.
The choice of appropriate endpoints is crucial and should
be addressed in future research. Similarly, determining
optimal time for assessing primary endpoint and the mini-
mum duration of clinical trial/follow-up has to be further
investigated (expert consensus has suggested a minimum of
12 to 24 months). Moreover, patient-reported outcome
measures and side effects need to be incorporated. Recom-
mendations from the 2012 guideline that should be revisited
are outlined in Supplementary Table S4.
CONCLUSIONS
Since the first KDIGO GN guideline published in 2012,
important progress has been made in defining diseases
Table 5 | Examples of various rituximab-based regimens for
induction and remission in AAV that have been used in the
literature
Induction
Four weekly i.v. doses of 375 mg/m
2
,
171,172
or 2 biweekly doses of 750 mg/
m
2
(maximum dose 1000 mg)
182
Four weekly i.v. doses of 375 mg/m
2
and 1 monthly infusion 1 and 2
months apart
179,186
Maintenance
750 mg/m
2
(maximum dose 1000 mg) every 6 months
180–183
750 mg/m
2
(maximum dose 1000 mg) every 4 months
181
750 mg/m
2
(maximum dose 1000 mg) every 6 months for 24 months
184
750 mg/m
2
(maximum dose 1000 mg) every 12 months
183
375 mg/m
2
every 6 months
183
500 mg on days 1 and 15, then 5.5 months later, and again every 6
months for a total of 5 doses over 18 months
185
AAV, anti-neutrophil cytoplasmic antibody-associated vasculitis.
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
290 Kidney International (2019) 95, 281–295
(e.g., C3G), improving diagnostics (e.g., antiphospholipase
A2 receptor), identifying relevant biomarkers (e.g., DNA J
homolog subfamily B member 9), and applying new ther-
apies (e.g., rituximab in AAN). However, for any single
glomerular disease, we are still missing 1 or more crucial
pieces necessary for optimal clinical management. Con-
siderations around treatment futility and patient-centered
outcomes, which are important for all glomerular dis-
eases, are just emerging. This Controversies Conference
may be best summarized as an honest assessment of
where we are currently and a roadmap of where we need
to be.
APPENDIX
Other Conference Participants
Sharon G. Adler, USA; Charles E. Alpers, USA; Isabelle Ayoub, USA; Arvind
Bagga, India; Sean J. Barbour, Canada; Jonathan Barratt, UK; Daniel T.M.
Chan, Hong Kong; Anthony Chang, USA; Jason Chon Jun Choo, Singapore;
H. Terence Cook, UK; Rosanna Coppo, Italy; Fernando C. Fervenza, USA;
AgnesB.Fogo,USA;JonathanG.Fox,UK;RichardJ.Glassock,USA;David
Harris, Australia; Elisabeth M. Hodson, Australia; Jonathan J. Hogan, USA;
Elion Hoxha, Germany; Kunitoshi Iseki, Japan; J. Charles Jennette, USA;
Vivekanand Jha, India; David W. Johnson, Australia; Shinya Kaname, Japan;
Ritsuko Katafuchi, Japan; A. Richard Kitching, Australia; Richard A.
Lafayette, USA; Philip K.T. Li, Hong Kong; Adrian Liew, Singapore; Jicheng
Lv, China; Ana Malvar, Argentina; Shoichi Maruyama, Japan; Juan Manuel
Mejía-Vilet, Mexico; Chi Chiu Mok, Hong Kong; Patrick H. Nachman, USA;
Carla M. Nester, USA; Eisei Noiri, Japan; Michelle M. O’Shaughnessy, USA;
Seza Özen, Turkey; Samir M. Parikh, USA; Hyeong-Cheon Park, Korea; Chen
Au Peh, Australia; William F. Pendergraft, USA; Matthew C. Pickering, UK;
Evangéline Pillebout, France; Jai Radhakrishnan, USA; Manish Rathi, India;
Pierre Ronco, France; William E. Smoyer, USA; Sydney C.W. Tang, Hong
Kong; Vladimír Tesa
r, Czech Republic; Joshua M. Thurman, USA; Hernán
Trimarchi, Argentina; Marina Vivarelli, Italy; Giles D. Walters, Australia;
Angela Yee-Moon Wang, Hong Kong; ScottE.Wenderfer,USA;JackF.M.
Wetzels, The Netherlands.
SUPPLEMENTARY MATERIAL
Table S1. 2012 Kidney Disease: Improving Global Outcomes (KDIGO)
glomerulonephritis (GN) guideline recommendations related to
minimal change disease, focal segmental glomerulosclerosis (FSGS),
steroid-sensitive nephrotic syndrome (SSNS), and steroid-resistant
nephrotic syndrome (SRNS): Need to be revisited?
Table S2. 2012 Kidney Disease: Improving Global Outcomes (KDIGO)
glomerulonephritis (GN) guideline recommendations related to
idiopathic membranoproliferative glomerulonephritis: Need to be
revisited?
Table S3. 2012 Kidney Disease: Improving Global Outcomes (KDIGO)
glomerulonephritis (GN) guideline recommendations related to lupus
nephritis: Need to be revisited?
Table S4. 2012 Kidney Disease: Improving Global Outcomes (KDIGO)
glomerulonephritis (GN) guideline recommendations related to anti-
neutrophil cytoplasmic antibody–associated vasculitis (AAV): Need to
be revisited?
Supplementary material is linked to the online version of the paper at
www.kidney-international.org.
REFERENCES
1. Kidney Disease: Improving Global Outcomes (KDIGO)
Glomerulonephritis Work Group. KDIGO clinical practice guideline for
glomerulonephritis. Kidney Int Suppl. 2012;2:139–274.
2. Corwin HL, Schwartz MM, Lewis EJ. The importance of sample
size in the interpretation of the renal biopsy. Am J Nephrol.
1988;8:85–89.
3. Vivarelli M, Massella L, Ruggiero B, et al. Minimal change disease. Clin J
Am Soc Nephrol. 2017;12:332–345.
4. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am
Soc Nephrol. 2017;12:502–517.
5. De Vriese AS, Sethi S, Nath KA, et al. Differentiating primary, genetic,
and secondary FSGS in adults: a clinicopathologic approach. J Am Soc
Nephrol. 2018;29:759–774.
6. Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell
function. Lancet. 1974;2:556–560.
7. Maas RJ, Deegens JK, Wetzels JF. Permeability factors in idiopathic
nephrotic syndrome: historical perspectives and lessons for the future.
Nephrol Dial Transplant. 2014;29:2207–2216.
8. Hayek SS, Sever S, Ko YA, et al. Soluble urokinase receptor and chronic
kidney disease. N Engl J Med. 2015;373:1916–1925.
9. Clement LC, Avila-Casado C, Mace C, et al. Podocyte-secreted
angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive
nephrotic syndrome. Nat Med. 2011;17:117–122.
10. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in
idiopathic nephrotic syndrome and focal segmental glomerulosclerosis.
Clin J Am Soc Nephrol. 2010;5:2115–2121.
11. Shin JI, Kronbichler A. Rituximab for patients with nephrotic syndrome.
Lancet. 2015;385:225–226.
12. Novelli R, Gagliardini E, Ruggiero B, et al. Any value of podocyte B7-1 as
a biomarker in human MCD and FSGS? Am J Physiol Renal Physiol.
2016;310:F335–F341.
13. Shankland SJ, Smeets B, Pippin JW, et al. The emergence of the
glomerular parietal epithelial cell. Nat Rev Nephrol. 2014;10:158–
173.
14. D’Agati VD, Fogo AB, Bruijn JA, et al. Pathologic classification of focal
segmental glomerulosclerosis: a working proposal. Am J Kidney Dis.
2004;43:368–382.
15. D’Agati VD, Alster JM, Jennette JC, et al. Association of histologic
variants in FSGS clinical trial with presenting features and outcomes.
Clin J Am Soc Nephrol. 2013;8:399–406.
16. Smeets B, Stucker F, Wetzels J, et al. Detection of activated parietal
epithelial cells on the glomerular tuft distinguishes early focal
DISCLOSURE
BHR declared having received consultancy fees from Alexion, Aurinia, Biogen,
Biomarin, Bristol-Myers Squibb, ChemoCentryx, EMD Serono, Frazier Life
Sciences, Genentech, Gilead, Lupus Foundation of America, Mallinckrodt,
MedImmune, Novartis, Pharmalink, Ra Pharmaceuticals, Retrophin, and Rigel;
and travel support from American Society of Nephrology, Aurinia, Biogen,
Budapest Nephrology School, Childhood Arthritis and Rheumatology Research
Alliance, Chemocentryx, Congress on SLE (Australia), Central Society for Clinical
and Translational Research-Midwestern American Federation for Medical
Research, CureGN, European League Against Rheumatism Congress and
Portuguese Congress, KDIGO, MENTOR (Multicenter Randomized Controlled
Trial of Rituximab), Office of Minority Health Impact for Lupus, Pharmalink, Ra
Pharmaceuticals, Retrophin, and UpToDate. DJC declared having received
research support from National Institutes of Health. DCC declared having
received consultancy fees from Alnylam, Calliditas, ChemoCentryx, Dimerix,
Mallinckrodt, Novartis, and Rigel; and research support from Genentech and
National Institute of Diabetes, Digestive, and Kidney Diseases. KLG declared
having served on the chronic kidney disease advisory board of Reata. JJH
declared having received consultancy fees from Aurinia, Dimerix, and Variant.
MJM declared having received research support from German Ministry for
Science and Education (BMBF) and German Research Foundation (DFG). DCW
declared having received consultancy fees from Akebia, AstraZeneca, Amgen,
Boehringer Ingelheim, GlaxoSmithKline, Janssen, and Vifor Fresenius; speaker
honoraria from Amgen and Vifor Fresenius; and research support from
AstraZeneca. WCW declared having received consultancy fees from Akebia,
AMAG, Amgen, AstraZeneca, Bayer, Daichii-Sankyo, Relypsa, and ZS Pharma;
speaker honoraria from FibroGen; and research support from National
Institutes of Health. JF declared having received consultancy fees from Amgen,
Alnylam, Bayer, Boehringer Ingelheim, Calliditas, Inositec, Novo Nordisk,
Omeros, and Vifor; speaker honoraria from Amgen and Vifor; and travel
support from Boehringer Ingelheim. All other authors declared no competing
interests.
ACKNOWLEDGMENTS
The conference was sponsored by KDIGO and supported in part by
unrestricted educational grants from Achillion, Aurinia
Pharmaceuticals, Calliditas Therapeutics, ChemoCentryx, Chugai,
Expedition Therapeutics, Gilead, Goldfinch Bio, Kyowa Kirin,
Mallinckrodt Pharmaceuticals, Novartis, Omeros, SanofiGenzyme, and
Vifor Fresenius Medical Care Renal Pharma.
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 291
segmental glomerulosclerosis from minimal change disease. Am J
Pathol. 2014;184:3239–3248.
17. Gbadegesin RA, Winn MP, Smoyer WE. Genetic testing in nephrotic
syndrome—challenges and opportunities. Nat Rev Nephrol. 2013;9:
179–184.
18. Lovric S, Ashraf S, Tan W, et al. Genetic testing in steroid-resistant
nephrotic syndrome: when and how? Nephrol Dial Transplant. 2016;31:
1802–1813.
19. Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of
cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol.
2015;26:1279–1289.
20. Trautmann A, Schnaidt S, Lipska-Zietkiewicz BS, et al. Long-term
outcome of steroid-resistant nephrotic syndrome in children. J Am Soc
Nephrol. 2017;28:3055–3065.
21. Trautmann A, Bodria M, Ozaltin F, et al. Spectrum of steroid-resistant
and congenital nephrotic syndrome in children: the PodoNet registry
cohort. Clin J Am Soc Nephrol. 2015;10:592–600.
22. Teeninga N, Kist-van Holthe JE, van Rijswijk N, et al. Extending
prednisolone treatment does not reduce relapses in childhood
nephrotic syndrome. J Am Soc Nephrol. 2013;24:149–159.
23. Sinha A, Saha A, Kumar M, et al. Extending initial prednisolone
treatment in a randomized control trial from 3 to 6 months did not
significantly influence the course of illness in children with steroid-
sensitive nephrotic syndrome. Kidney Int. 2015;87:217–224.
24. Yoshikawa N, Nakanishi K, Sako M, et al. A multicenter randomized trial
indicates initial prednisolone treatment for childhood nephrotic
syndrome for two months is not inferior to six-month treatment. Kidney
Int. 2015;87:225–232.
25. Yadav M, Sinha A, Hari P, Bagga A. Efficacy of low-dose daily versus
alternate day prednisone in children with frequently relapsing
nephrotic syndrome (FRNS): open-label randomized controlled trial
(RCT). Abstract FP-S25-09. Pediatr Nephrol. 2016;31:1752.
26. Iijima K, Sako M, Nozu K, et al. Rituximab for childhood-onset,
complicated, frequently relapsing nephrotic syndrome or steroid-
dependent nephrotic syndrome: a multicentre, double-blind,
randomised, placebo-controlled trial. Lancet. 2014;384:1273–1281.
27. Kim AH, Chung JJ, Akilesh S, et al. B cell-derived IL-4 acts on podocytes
to induce proteinuria and foot process effacement. JCI Insight. 2017;2.
pii:81836.
28. Gellermann J, Weber L, Pape L, et al. Mycophenolate mofetil versus
cyclosporin A in children with frequently relapsing nephrotic
syndrome. J Am Soc Nephrol. 2013;24:1689–1697.
29. Masse V, Al Jijakli A, Genet P, et al. Screening and management of
hepatitis B virus before the first rituximab infusion: We must do better!
Blood. 2014;124:2754. Available at: http://www.bloodjournal.org/
content/124/21/2754. Accessed March 15, 2018.
30. Sethi S, Haas M, Markowitz GS, et al. Mayo Clinic/Renal Pathology
Society Consensus report on pathologic classification, diagnosis, and
reporting of GN. J Am Soc Nephrol. 2016;27:1278–1287.
31. Fervenza FC, Sethi S, Glassock RJ. Idiopathic membranoproliferative
glomerulonephritis: does it exist? Nephrol Dial Transplant. 2012;27:
4288–4294.
32. Nasr SH, Galgano SJ, Markowitz GS, et al. Immunofluorescence on
pronase-digested paraffin sections: a valuable salvage technique for
renal biopsies. Kidney Int. 2006;70:2148–2151.
33. Larsen CP, Ambuzs JM, Bonsib SM, et al. Membranous-like
glomerulopathy with masked IgG kappa deposits. Kidney Int. 2014;86:
154–161.
34. Messias NC, Walker PD, Larsen CP. Paraffin immunofluorescence in the
renal pathology laboratory: more than a salvage technique. Mod Pathol.
2015;28:854–860.
35. Larsen CP, Messias NC, Walker PD, et al. Membranoproliferative
glomerulonephritis with masked monotypic immunoglobulin deposits.
Kidney Int. 2015;88:867–873.
36. Sethi S, Nasr SH, De Vriese AS, et al. C4d as a diagnostic tool in
proliferative GN. J Am Soc Nephrol. 2015;26:2852–2859.
37. Andeen NK, Yang HY, Dai DF, et al. DnaJ homolog subfamily B member
9 is a putative autoantigen in fibrillary GN. J Am Soc Nephrol. 2018;29:
231–239.
38. Dasari S, Alexander MP, Vrana JA, et al. DnaJ heat shock protein family B
member 9 is a novel biomarker for fibrillary GN. J Am Soc Nephrol.
2018;29:51–56.
39. Pickering MC, D’Agati VD, Nester CM, et al. C3 glomerulopathy:
consensus report. Kidney Int. 2013;84:1079–1089.
40. Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic
syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease:
Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney
Int. 2017;91:539–551.
41. Servais A, Noel LH, Roumenina LT, et al. Acquired and genetic
complement abnormalities play a critical role in dense deposit disease
and other C3 glomerulopathies. Kidney Int. 2012;82:454–464.
42. Iatropoulos P, Daina E, Curreri M, et al. Cluster analysis identifies distinct
pathogenetic patterns in C3 glomerulopathies/immune complex-
mediated membranoproliferative GN. J Am Soc Nephrol. 2018;29:
283–294.
43. Zand L, Kattah A, Fervenza FC, et al. C3 glomerulonephritis associated
with monoclonal gammopathy: a case series. Am J Kidney Dis. 2013;62:
506–514.
44. Avasare RS, Canetta PA, Bomback AS, et al. Mycophenolate mofetil in
combination with steroids for treatment of C3 glomerulopathy: a case
series. Clin J Am Soc Nephrol. 2018;13:406–413.
45. Rabasco C, Cavero T, Roman E, et al. Effectiveness of mycophenolate
mofetil in C3 glomerulonephritis. Kidney Int. 2015;88:1153–1160.
46. Caliskan Y, Torun ES, Tiryaki TO, et al. Immunosuppressive treatment in
C3 glomerulopathy: is it really effective? Am J Nephrol. 2017;46:96–107.
47. Chauvet S, Fremeaux-Bacchi V, Petitprez F, et al. Treatment of B-cell
disorder improves renal outcome of patients with monoclonal
gammopathy-associated C3 glomerulopathy. Blood. 2017;129:1437–1447.
48. Vignon M, Cohen C, Faguer S, et al. The clinicopathologic characteristics
of kidney diseases related to monotypic IgA deposits. Kidney Int.
2017;91:720–728.
49. Bonaud A, Bender S, Touchard G, et al. A mouse model recapitulating
human monoclonal heavy chain deposition disease evidences the
relevance of proteasome inhibitor therapy. Blood. 2015;126:757–765.
50. Bridoux F, Javaugue V, Bender S, et al. Unravelling the
immunopathological mechanisms of heavy chain deposition disease
with implications for clinical management. Kidney Int. 2017;91:423–434.
51. Cohen C, Royer B, Javaugue V, et al. Bortezomib produces high
hematological response rates with prolonged renal survival in
monoclonal immunoglobulin deposition disease. Kidney Int. 2015;88:
1135–1143.
52. Bridoux F, Leung N, Hutchison CA, et al. Diagnosis of monoclonal
gammopathy of renal significance. Kidney Int. 2015;87:698–711.
53. Gumber R, Cohen JB, Palmer MB, et al. A clone-directed approach may
improve diagnosis and treatment of proliferative glomerulonephritis
with monoclonal immunoglobulin deposits. Kidney Int. 2018;94:
199–205.
54. Bhutani G, Nasr SH, Said SM, et al. Hematologic characteristics of
proliferative glomerulonephritides with nonorganized monoclonal
immunoglobulin deposits. Mayo Clin Proc. 2015;90:587–596.
55. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working
Group consensus criteria for response and minimal residual disease
assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346.
56. Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the
conduct and reporting of clinical trials in systemic light-chain
amyloidosis. Leukemia. 2012;26:2317–2325.
57. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to
treatment in immunoglobulin light chain amyloidosis based on free
light chain measurement and cardiac biomarkers: impact on survival
outcomes. J Clin Oncol. 2012;30:4541–4549.
58. Dimopoulos MA, Roussou M, Gavriatopoulou M, et al. Outcomes of
newly diagnosed myeloma patients requiring dialysis: renal recovery,
importance of rapid response and survival benefit. Blood Cancer J.
2017;7:e571.
59. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal
outcome and early markers of renal response to chemotherapy in AL
amyloidosis. Blood. 2014;124:2325–2332.
60. Vignon M, Javaugue V, Alexander MP, et al. Current anti-myeloma
therapies in renal manifestations of monoclonal light chain-associated
Fanconi syndrome: a retrospective series of 49 patients. Leukemia.
2017;31:123–129.
61. Fermand JP, Bridoux F, Kyle RA, et al. How I treat monoclonal
gammopathy of renal significance (MGRS). Blood. 2013;122:3583–3590.
62. Sawinski D, Lim MA, Cohen JB, et al. Patient and kidney allograft
survival in recipients with end-stage renal disease from amyloidosis.
Transplantation. 2018;102:300–309.
63. Kidney Disease: Improving Global Outcomes (KDIGO) Hepatitis C Work
Group. KDIGO 2018 clinical practice guideline on the prevention,
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
292 Kidney International (2019) 95, 281–295
diagnosis, evaluation, and treatment of hepatitis c in chronic kidney
disease. Kidney Int Suppl. 2018;8:91–165.
64. Levine JW, Gota C, Fessler BJ, et al. Persistent cryoglobulinemic
vasculitis following successful treatment of hepatitis C virus.
J Rheumatol. 2005;32:1164–1167.
65. Landau DA, Saadoun D, Halfon P, et al. Relapse of hepatitis C virus-
associated mixed cryoglobulinemia vasculitis in patients with sustained
viral response. Arthritis Rheum. 2008;58:604–611.
66. Ghosn M, Palmer MB, Najem CE, et al. New-onset hepatitis C virus-
associated glomerulonephritis following sustained virologic response
with direct-acting antiviral therapy. Clin Nephrol. 2017;87:261–266.
67. Artemova M, Abdurakhmanov D, Ignatova T, et al. Persistent hepatitis C
virus-associated cryoglobulinemic vasculitis following virus eradication
after direct-acting antiviral therapy. Hepatology. 2017;65:1770–1771.
68. Hogan J, Restivo M, Canetta PA, et al. Rituximab treatment for fibrillary
glomerulonephritis. Nephrol Dial Transplant. 2014;29:1925–1931.
69. Javaugue V, Karras A, Glowacki F, et al. Long-term kidney disease
outcomes in fibrillary glomerulonephritis: a case series of 27 patients.
Am J Kidney Dis. 2013;62:679–690.
70. Kalbermatter SA, Marone C, Casartelli D, et al. Outcome of fibrillary
glomerulonephritis. Swiss Med Wkly. 2012;142:w13578.
71. Nasr SH, Valeri AM, Cornell LD, et al. Fibrillary glomerulonephritis: a
report of 66 cases from a single institution. Clin J Am Soc Nephrol.
2011;6:775–784.
72. Rosenstock JL, Markowitz GS, Valeri AM, et al. Fibrillary and
immunotactoid glomerulonephritis: distinct entities with different
clinical and pathologic features. Kidney Int. 2003;63:1450–1461.
73. Weening JJ, D’Agati VD, Schwartz MM, et al. The classification of
glomerulonephritis in systemic lupus erythematosus revisited. Kidney
Int. 2004;65:521–530.
74. Yu F, Wu LH, Tan Y, et al. Tubulointerstitial lesions of patients with
lupus nephritis classified by the 2003 International Society of
Nephrology and Renal Pathology Society system. Kidney Int. 2010;77:
820–829.
75. Wu LH, Yu F, Tan Y, et al. Inclusion of renal vascular lesions in the 2003
ISN/RPS system for classifying lupus nephritis improves renal outcome
predictions. Kidney Int. 2013;83:715–723.
76. Hu W, Chen Y, Wang S, et al. Clinical-morphological features and
outcomes of lupus podocytopathy. Clin J Am Soc Nephrol. 2016;11:
585–592.
77. Yu F, Haas M, Glassock R, et al. Redefining lupus nephritis: clinical
implications of pathophysiologic subtypes. Nat Rev Nephrol. 2017;13:
483–495.
78. Yu F, Tan Y, Liu G, et al. Clinicopathological characteristics and
outcomes of patients with crescentic lupus nephritis. Kidney Int.
2009;76:307–317.
79. Hsieh C, Chang A, Brandt D, et al. Predicting outcomes of lupus
nephritis with tubulointerstitial inflammation and scarring. Arthritis Care
Res. 2011;63:865–874.
80. Mejia-Vilet JM, Cordova-Sanchez BM, Uribe-Uribe NO, et al. Prognostic
significance of renal vascular pathology in lupus nephritis. Lupus.
2017;26:1042–1050.
81. Haring CM, Rietveld A, van den Brand JA, et al. Segmental and global
subclasses of class IV lupus nephritis have similar renal outcomes. JAm
Soc Nephrol. 2012;23:149–154.
82. Schwartz MM, Korbet SM, Lewis EJ, et al. The prognosis and
pathogenesis of severe lupus glomerulonephritis. Nephrol Dial
Transplant. 2008;23:1298–1306.
83. Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International
Society of Nephrology/Renal Pathology Society classification for lupus
nephritis: clarification of definitions, and modified National Institutes of
Health activity and chronicity indices. Kidney Int. 2018;93:789–796.
84. Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the
Systemic Lupus International Collaborating Clinics classification
criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:
2677–2686.
85. Rijnink EC, Teng YKO, Kraaij T, et al. Validation of the Systemic Lupus
International Collaborating Clinics classification criteria in a cohort of
patients with full house glomerular deposits. Kidney Int. 2018;93:
214–220.
86. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:
2110–2121.
87. Munroe ME, James JA. Genetics of lupus nephritis: clinical implications.
Semin Nephrol. 2015;35:396–409.
88. Caster DJ, Korte EA, Nanda SK, et al. ABIN1 dysfunction as a genetic
basis for lupus nephritis. J Am Soc Nephrol. 2013;24:1743–1754.
89. Bomback AS, Gharavi AG. Lupus nephritis: ancestry, genetic risk and
health disparities. Nat Rev Nephrol. 2013;9:699–700.
90. Ceccarelli F, Perricone C, Borgiani P, et al. Genetic factors in systemic
lupus erythematosus: contribution to disease phenotype. J Immunol
Res. 2015:745647.
91. Freedman BI, Langefeld CD, Andringa KK, et al. End-stage renal disease
in African Americans with lupus nephritis is associated with APOL1.
Arthritis Rheumatol. 2014;66:390–396.
92. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group.
KDIGO 2012 clinical practice guideline for the evaluation and
management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150.
93. Condon MB, Ashby D, Pepper RJ, et al. Prospective observational single-
centre cohort study to evaluate the effectiveness of treating lupus
nephritis with rituximab and mycophenolate mofetil but no oral
steroids. Ann Rheum Dis. 2013;72:1280–1286.
94. Malvar A, Pirruccio P, Alberton V, et al. Histologic versus clinical
remission in proliferative lupus nephritis. Nephrol Dial Transplant.
2017;32:1338–1344.
95. Parikh SV, Alvarado A, Malvar A, et al. The kidney biopsy in lupus
nephritis: past, present, and future. Semin Nephrol. 2015;35:465–477.
96. Alvarado AS, Malvar A, Lococo B, et al. The value of repeat kidney
biopsy in quiescent Argentinian lupus nephritis patients. Lupus.
2014;23:840–847.
97. Zickert A, Sundelin B, Svenungsson E, et al. Role of early repeated renal
biopsies in lupus nephritis. Lupus Sci Med. 2014;1:e000018.
98. Dall’Era M, Cisternas MG, Smilek DE, et al. Predictors of long-term
renal outcome in lupus nephritis trials: lessons learned from the
Euro-Lupus Nephritis cohort. Arthritis Rheumatol. 2015;67:
1305–1313.
99. Tamirou F, D’Cruz D, Sangle S, et al. Long-term follow-up of the
MAINTAIN Nephritis Trial, comparing azathioprine and mycophenolate
mofetil as maintenance therapy of lupus nephritis. Ann Rheum Dis.
2016;75:526–531.
100. Ugolini-Lopes MR, Seguro LPC, Castro MXF, et al. Early proteinuria
response: a valid real-life situation predictor of long-term lupus renal
outcome in an ethnically diverse group with severe biopsy-proven
nephritis? Lupus Sci Med. 2017;4:e000213.
101. Birmingham DJ, Shidham G, Perna A, et al. Spot PC ratio estimates of
24-hour proteinuria are more unreliable in lupus nephritis than in other
forms of chronic glomerular disease. Ann Rheum Dis. 2014;73:475–476.
102. Yang XW, Tan Y, Yu F, et al. Combination of anti-C1q and anti-dsDNA
antibodies is associated with higher renal disease activity and predicts
renal prognosis of patients with lupus nephritis. Nephrol Dial Transplant.
2012;27:3552–3559.
103. Orbai AM, Truedsson L, Sturfelt G, et al. Anti-C1q antibodies in systemic
lupus erythematosus. Lupus. 2015;24:42–49.
104. Soliman S, Mohan C. Lupus nephritis biomarkers. Clin Immunol.
2017;185:10–20.
105. Phatak S, Chaurasia S, Mishra SK, et al. Urinary B cell activating factor
(BAFF) and a proliferation-inducing ligand (APRIL): potential biomarkers
of active lupus nephritis. Clin Exp Immunol. 2017;187:376–382.
106. Xuejing Z, Jiazhen T, Jun L, et al. Urinary TWEAK level as a marker of
lupus nephritis activity in 46 cases. J Biomed Biotechnol. 2012;2012:
359647.
107. Reyes-Thomas J, Blanco I, Putterman C. Urinary biomarkers in lupus
nephritis. Clin Rev Allergy Immunol. 2011;40:138–150.
108. Parikh SV, Malvar A, Song H, et al. Molecular imaging of the kidney in
lupus nephritis to characterize response to treatment. Transl Res.
2017;182:1–13.
109. Parikh SV, Malvar A, Song H, et al. Characterising the immune profile of
the kidney biopsy at lupus nephritis flare differentiates early treatment
responders from non-responders. Lupus Sci Med. 2015;2:e000112.
110. Banchereau R, Hong S, Cantarel B, et al. Personalized
immunomonitoring uncovers molecular networks that stratify lupus
patients. Cell. 2016;165:551–565.
111. Pons-Estel GJ, Alarcon GS, Burgos PI, et al. Mestizos with systemic lupus
erythematosus develop renal disease early while antimalarials retard its
appearance: data from a Latin American cohort. Lupus. 2013;22:
899–907.
112. Ugarte-Gil MF, Wojdyla D, Pastor-Asurza CA, et al. Predictive factors of
flares in systemic lupus erythematosus patients: data from a
multiethnic Latin American cohort. Lupus. 2018;27:536–544.
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 293
113. Fessler BJ, Alarcon GS, McGwin G Jr, et al. Systemic lupus
erythematosus in three ethnic groups: XVI. Association of
hydroxychloroquine use with reduced risk of damage accrual. Arthritis
Rheum. 2005;52:1473–1480.
114. Galindo-Izquierdo M, Rodriguez-Almaraz E, Pego-Reigosa JM, et al.
Characterization of patients with lupus nephritis included in a large
cohort from the Spanish Society of Rheumatology Registry of Patients
With Systemic Lupus Erythematosus (RELESSER). Medicine. 2016;95:
e2891.
115. Joo YB, Won S, Choi CB, et al. Lupus nephritis is associated with more
corticosteroid-associated organ damage but less corticosteroid non-
associated organ damage. Lupus. 2017;26:598–605.
116. Sciascia S, Mompean E, Radin M, et al. Rate of adverse effects of
medium- to high-dose glucocorticoid therapy in systemic lupus
erythematosus: a systematic review of randomized control trials. Clin
Drug Investig. 2017;37:519–524.
117. Roccatello D, Sciascia S, Baldovino S, et al. A 4-year observation in lupus
nephritis patients treated with an intensified B-lymphocyte depletion
without immunosuppressive maintenance treatment: clinical response
compared to literature and immunological re-assessment. Autoimmun
Rev. 2015;14:1123–1130.
118. Ruiz-Irastorza G, Ugarte A, Saint-Pastou Terrier C, et al. Repeated pulses
of methyl-prednisolone with reduced doses of prednisone improve the
outcome of class III, IV and V lupus nephritis: an observational
comparative study of the Lupus-Cruces and lupus-Bordeaux cohorts.
Autoimmun Rev. 2017;16:826–832.
119. Liu Z, Zhang H, Liu Z, et al. Multitargettherapyforinduction
treatment of lupus nephritis: a randomized trial. AnnInternMed.
2015;162:18–26.
120. Zhang H, Liu Z, Zhou M, et al. Multitarget therapy for maintenance
treatment of lupus nephritis. J Am Soc Nephrol. 2017;28:3671–3678.
121. Dooley MA, Jayne D, Ginzler EM, et al. Mycophenolate versus
azathioprine as maintenance therapy for lupus nephritis. N Engl J Med.
2011;365:1886–1895.
122. Houssiau FA, D’Cruz D, Sangle S, et al. Azathioprine versus
mycophenolate mofetil for long-term immunosuppression in lupus
nephritis: results from the MAINTAIN Nephritis Trial. Ann Rheum Dis.
2010;69:2083–2089.
123. Lenz O, Waheed AA, Baig A, et al. Lupus nephritis: maintenance therapy
for lupus nephritis: Do we now have a plan? Clin J Am Soc Nephrol.
2013;8:162–171.
124. Moroni G, Gallelli B, Quaglini S, et al. Withdrawal of therapy in patients
with proliferative lupus nephritis: long-term follow-up. Nephrol Dial
Transplant. 2006;21:1541–1548.
125. Grootscholten C, Berden JH. Discontinuation of immunosuppression in
proliferative lupus nephritis: is it possible? Nephrol Dial Transplant.
2006;21:1465–1469.
126. Ioannidis JP, Boki KA, Katsorida ME, et al. Remission, relapse, and re-
remission of proliferative lupus nephritis treated with
cyclophosphamide. Kidney Int. 2000;57:258–264.
127. Mok CC, Ying KY, Tang S, et al. Predictors and outcome of renal flares
after successful cyclophosphamide treatment for diffuse proliferative
lupus glomerulonephritis. Arthritis Rheum. 2004;50:2559–2568.
128. Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in
patients with active proliferative lupus nephritis: the Lupus Nephritis
Assessment with Rituximab study. Arthritis Rheum. 2012;64:1215–1226.
129. Duxbury B, Combescure C, Chizzolini C. Rituximab in systemic lupus
erythematosus: an updated systematic review and meta-analysis. Lupus.
2013;22:1489–1503.
130. Shamliyan TA, Dospinescu P. Additional improvements in clinical
response from adjuvant biologic response modifiers in adults with
moderate to severe systemic lupus erythematosus despite
immunosuppressive agents: a systematic review and meta-analysis. Clin
Ther. 2017;39:1479–1506.
131. Moroni G, Raffiotta F, Trezzi B, et al. Rituximab vs mycophenolate and vs
cyclophosphamide pulses for induction therapy of active lupus
nephritis: a clinical observational study. Rheumatology. 2014;53:
1570–1577.
132. Mok CC, Yap DY, Navarra SV, et al. Overview of lupus nephritis
management guidelines and perspective from Asia. Int J Rheum Dis.
2013;16:625–636.
133. Chavarot N, Verhelst D, Pardon A, et al. Rituximab alone as induction
therapy for membranous lupus nephritis: a multicenter retrospective
study. Medicine. 2017;96:e7429.
134. Song D, Wu LH, Wang FM, et al. The spectrum of renal thrombotic
microangiopathy in lupus nephritis. Arthritis Res Ther. 2013;15:R12.
135. Pattanashetti N, Anakutti H, Ramachandran R, et al. Effect of thrombotic
microangiopathy on clinical outcomes in Indian patients with lupus
nephritis. Kidney Int Rep. 2017;2:844–849.
136. Chen MH, Chen MH, Chen WS, et al. Thrombotic microangiopathy in
systemic lupus erythematosus: a cohort study in North Taiwan.
Rheumatology. 2011;50:768–775.
137. Kronbichler A, Brezina B, Quintana LF, et al. Efficacy of plasma exchange
and immunoadsorption in systemic lupus erythematosus and
antiphospholipid syndrome: a systematic review. Autoimmun Rev.
2016;15:38–49.
138. Sciascia S, Radin M, Yazdany J, et al. Expanding the therapeutic options
for renal involvement in lupus: eculizumab, available evidence.
Rheumatol Int. 2017;37:1249–1255.
139. de Holanda MI, Porto LC, Wagner T, et al. Use of eculizumab in a
systemic lupus erythemathosus patient presenting thrombotic
microangiopathy and heterozygous deletion in CFHR1-CFHR3: a case
report and systematic review. Clin Rheumatol. 2017;36:2859–2867.
140. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor
eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med.
2013;368:2169–2181.
141. Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab
in atypical hemolytic uremic syndrome from 2-year extensions of phase
2 studies. Kidney Int. 2015;87:1061–1073.
142. Erkan D, Aguiar CL, Andrade D, et al. 14th International Congress on
Antiphospholipid Antibodies: task force report on antiphospholipid
syndrome treatment trends. Autoimmun Rev. 2014;13:685–696.
143. Bienaime F, Legendre C, Terzi F, et al. Antiphospholipid syndrome and
kidney disease. Kidney Int. 2017;91:34–44.
144. Canaud G, Bienaime F, Tabarin F, et al. Inhibition of the mTORC
pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371:
303–312.
145. Webster P, Wardle A, Bramham K, et al. Tacrolimus is an effective
treatment for lupus nephritis in pregnancy. Lupus. 2014;23:1192–1196.
146. O’Shaughnessy MM, Liu S, Montez-Rath ME, et al. Kidney
transplantation outcomes across GN subtypes in the United States.
J Am Soc Nephrol. 2017;28:632–644.
147. Yu TM, Chen YH, Lan JL, et al. Renal outcome and evolution of disease
activity in Chinese lupus patients after renal transplantation. Lupus.
2008;17:687–694.
148. Yu TM, Wen MC, Li CY, et al. Impact of recurrent lupus nephritis on
lupus kidney transplantation: a 20-year single center experience. Clin
Rheumatol. 2012;31:705–710.
149. Naranjo-Escobar J, Manzi E, Posada JG, et al. Kidney transplantation for
end-stage renal disease in lupus nephritis, a very safe procedure: a
single Latin American transplant center experience. Lupus. 2017;26:
1157–1165.
150. Contreras G, Mattiazzi A, Guerra G, et al. Recurrence of lupus nephritis
after kidney transplantation. J Am Soc Nephrol. 2010;21:1200–1207.
151. Briganti EM, Russ GR, McNeil JJ, et al. Risk of renal allograft loss from
recurrent glomerulonephritis. N Engl J Med. 2002;347:103–109.
152. Mina R, von Scheven E, Ardoin SP, et al. Consensus treatment plans for
induction therapy of newly diagnosed proliferative lupus nephritis in
juvenile systemic lupus erythematosus. Arthritis Care Res. 2012;64:375–
383.
153. Hugle B, Silverman ED, Tyrrell PN, et al. Presentation and outcome of
paediatric membranous non-proliferative lupus nephritis. Pediatr
Nephrol. 2015;30:113–121.
154. Pereira M, Muscal E, Eldin K, et al. Clinical presentation and outcomes of
childhood-onset membranous lupus nephritis. Pediatr Nephrol. 2017;32:
2283–2291.
155. Groot N, de Graeff N, Marks SD, et al. European evidence-based
recommendations for the diagnosis and treatment of childhood-onset
lupus nephritis: the SHARE initiative. Ann Rheum Dis. 2017;76:
1965–1973.
156. Jennette JC, Nachman PH. ANCA glomerulonephritis and vasculitis. Clin
J Am Soc Nephrol. 2017;12:1680–1691.
157. Roth AJ, Ooi JD, Hess JJ, et al. Epitope specificity determines
pathogenicity and detectability in ANCA-associated vasculitis. J Clin
Invest. 2013;123:1773–1783.
158. Lionaki S, Blyth ER, Hogan SL, et al. Classification of antineutrophil
cytoplasmic autoantibody vasculitides: the role of antineutrophil
cytoplasmic autoantibody specificity for myeloperoxidase or proteinase
KDIGO executive conclusions BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report
294 Kidney International (2019) 95, 281–295
3 in disease recognition and prognosis. Arthritis Rheum. 2012;64:
3452–3462.
159. Walsh M, Flossmann O, Berden A, et al. Risk factors for relapse of
antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis
Rheum. 2012;64:542–548.
160. Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within
ANCA-associated vasculitis. N Engl J Med. 2012;367:214–223.
161. Schreiber A, Xiao H, Jennette JC, et al. C5a receptor mediates neutrophil
activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol.
2009;20:289–298.
162. Caster DJ, Powell DW, Miralda I, et al. Re-examining neutrophil
participation in GN. J Am Soc Nephrol. 2017;28:2275–2289.
163. Diaz-Crespo F, Villacorta J, Acevedo M, et al. The predictive value of
kidney biopsy in renal vasculitis: a multicenter cohort study. Hum
Pathol. 2016;52:119–127.
164. Tomasson G, Grayson PC, Mahr AD, et al. Value of ANCA measurements
during remission to predict a relapse of ANCA-associated vasculitis: a
meta-analysis. Rheumatology. 2012;51:100–109.
165. Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the
Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis.
2009;68:1827–1832.
166. Exley AR, Bacon PA, Luqmani RA, et al. Development and initial
validation of the Vasculitis Damage Index for the standardized clinical
assessment of damage in the systemic vasculitides. Arthritis Rheum.
1997;40:371–380.
167. Monach PA, Warner RL, Tomasson G, et al. Serum proteins reflecting
inflammation, injury and repair as biomarkers of disease activity in
ANCA-associated vasculitis. Ann Rheum Dis. 2013;72:1342–1350.
168. Ishizaki J, Takemori A, Suemori K, et al. Targeted proteomics reveals
promising biomarkers of disease activity and organ involvement in
antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res
Ther. 2017;19:218.
169. O’Reilly VP, Wong L, Kennedy C, et al. Urinary soluble CD163 in active
renal vasculitis. J Am Soc Nephrol. 2016;27:2906–2916.
170. Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a
receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc
Nephrol. 2017;28:2756–2767.
171. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus
cyclophosphamide for ANCA-associated vasculitis. N Engl J Med.
2010;363:221–232.
172. Specks U, Merkel PA, Seo P, et al. Efficacy of remission-induction
regimens for ANCA-associated vasculitis. N Engl J Med. 2013;369:
417–427.
173. Geetha D, Specks U, Stone JH, et al. Rituximab versus
cyclophosphamide for ANCA-associated vasculitis with renal
involvement. J Am Soc Nephrol. 2015;26:976–985.
174. Unizony S, Villarreal M, Miloslavsky EM, et al. Clinical outcomes of
treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated
vasculitis based on ANCA type. Ann Rheum Dis. 2016;75:1166–1169.
175. Miloslavsky EM, Lu N, Unizony S, et al. Myeloperoxidase-antineutrophil
cytoplasmic antibody (ANCA)-positive and ANCA-negative patients
with granulomatosis with polyangiitis (Wegener’s): distinct patient
subsets. Arthritis Rheumatol. 2016;68:2945–2952.
176. Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus
cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med.
2010;363:211–220.
177. Jones RB, Furuta S, Tervaert JW, et al. Rituximab versus
cyclophosphamide in ANCA-associated renal vasculitis: 2-year results of
a randomised trial. Ann Rheum Dis. 2015;74:1178–1182.
178. Karras A, Pagnoux C, Haubitz M, et al. Randomised controlled trial of
prolonged treatment in the remission phase of ANCA-associated
vasculitis. Ann Rheum Dis. 2017;76:1662–1668.
179. Roccatello D, Sciascia S, Rossi D, et al. The "4 plus 2" rituximab protocol
makes maintenance treatment unneeded in patients with refractory
ANCA-associated vasculitis: a 10 years observation study. Oncotarget.
2017;8:52072–52077.
180. Jones RB, Ferraro AJ, Chaudhry AN, et al. A multicenter survey of
rituximab therapy for refractory antineutrophil cytoplasmic antibody-
associated vasculitis. Arthritis Rheum. 2009;60:2156–2168.
181. Rhee EP, Laliberte KA, Niles JL. Rituximab as maintenance therapy for
anti-neutrophil cytoplasmic antibody-associated vasculitis. Clin J Am
Soc Nephrol. 2010;5:1394–1400.
182. Cartin-Ceba R, Golbin JM, Keogh KA, et al. Rituximab for remission
induction and maintenance in refractory granulomatosis with
polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis
Rheum. 2012;64:3770–3778.
183. Roubaud-Baudron C, Pagnoux C, Meaux-Ruault N, et al. Rituximab
maintenance therapy for granulomatosis with polyangiitis and
microscopic polyangiitis. J Rheumatol. 2012;39:125–130.
184. Smith RM, Jones RB, Guerry MJ, et al. Rituximab for remission
maintenance in relapsing antineutrophil cytoplasmic antibody-
associated vasculitis. Arthritis Rheum. 2012;64:3760–3769.
185. Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for
maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371:
1771–1780.
186. Roccatello D. How I treat" autoimmune diseases: state of the art on the
management of rare rheumatic diseases and ANCA-associated systemic
idiopathic vasculitis. Autoimmun Rev. 2017;16:995–998.
187. Wilkinson NM, Page J, Uribe AG, et al. Establishment of a pilot pediatric
registry for chronic vasculitis is both essential and feasible: a Childhood
Arthritis and Rheumatology Alliance (CARRA) survey. J Rheumatol.
2007;34:224–226.
188. Cabral DA, Canter DL, Muscal E, et al. Comparing presenting clinical
features in 48 children with microscopic polyangiitis to 183 children
who have granulomatosis with polyangiitis (Wegener’s): an ARChiVe
Cohort Study. Arthritis Rheumatol. 2016;68:2514–2526.
189. Eleftheriou D, Melo M, Marks SD, et al. Biologic therapy in
primary systemic vasculitis of the young. Rheumatology. 2009;48:
978–986.
190. James KE, Xiao R, Merkel PA, et al. Clinical course and outcomes of
childhood-onset granulomatosis with polyangiitis. Clin Exp Rheumatol.
2017;35(suppl 103):202–208.
BH Rovin et al.: Management and treatment of GN (part 2): a KDIGO conference report KDIGO executive conclusions
Kidney International (2019) 95, 281–295 295