Figure 5 - uploaded by Daniela T Pilz
Content may be subject to copyright.

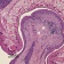
Cobblestone lissencephaly (US scan). Coronal ultrasound section. Falx (small arrows), lateral ventricle (LV), cisterna magna (CM). Smooth agyric cortex (medium arrows), hypoplastic cerebellum (large arrow). Hypoplastic inferior vermis seen on sagittal sections.
Source publication






Earl Walker's paper in 1942 represents a detailed review of early described cases of lissencephaly and states that Owen is said to have introduced the term lissencephaly to describe an agyric brain, from the Greek words 'lissos' (smooth) and 'encephalus' (brain). Further reports of lissencephaly followed by Miller, Dieker et al, Warburg, and others...
Similar publications




Congenital muscular dystrophies (CMD) are autosomal recessive, heterogeneous disorders. The commonest forms are the Fukuyama CMD (FCMD), associated with mental retardation and structural brain anomalies, and classical (occidental) CMD, with pure muscle expression. FCMD has been localized to chromosome 9q31 – q33. Following the discovery of merosin...
Citations
... 18 Mutations in Crk and Crkl are associated with several human diseases. [19][20][21][22][23][24] Importantly, Crkl is localized to 22q11.2, which is hemizygously deleted in the 22q11.2 deletion syndrome (22q11.2DS), ...

Background
Endocardial cells are a major progenitor population that gives rise to heart valves through endocardial cushion formation by endocardial to mesenchymal transformation and the subsequent endocardial cushion remodeling. Genetic variants that affect these developmental processes can lead to congenital heart valve defects. Crk and Crkl are ubiquitously expressed genes encoding cytoplasmic adaptors essential for cell signaling. This study aims to explore the specific role of Crk and Crkl in the endocardial lineage during heart valve development.
Methods and Results
We deleted Crk and Crkl specifically in the endocardial lineage. The resultant heart valve morphology was evaluated by histological analysis, and the underlying cellular and molecular mechanisms were investigated by immunostaining and quantitative reverse transcription polymerase chain reaction. We found that the targeted deletion of Crk and Crkl impeded the remodeling of endocardial cushions at the atrioventricular canal into the atrioventricular valves. We showed that apoptosis was temporally increased in the remodeling atrioventricular endocardial cushions, and this developmentally upregulated apoptosis was repressed by deletion of Crk and Crkl . Loss of Crk and Crkl also resulted in altered extracellular matrix production and organization in the remodeling atrioventricular endocardial cushions. These morphogenic defects were associated with altered expression of genes in BMP (bone morphogenetic protein), connective tissue growth factor, and WNT signaling pathways, and reduced extracellular signal‐regulated kinase signaling activities.
Conclusions
Our findings support that Crk and Crkl have shared functions in the endocardial lineage that critically regulate atrioventricular valve development; together, they likely coordinate the morphogenic signals involved in the remodeling of the atrioventricular endocardial cushions.
... RBP TRIM71 mutations in humans and homozygous deletion in mice affect neurogenesis-dependent biomechanics causing monogenic hydrocephalus and ventricular CSF-parenchyma defects in the absence of altered CSF flow (Duy et al. 2022). LIS1 mutations in humans causing lissencephaly or MDLS show typical features of ventriculomegaly or hydrocephalus (Holzgreve et al. 1993;Pilz and Quarrell 1996;Zhang et al. 2022). Moreover, heterozygous Lis1 hypomorphs and conditional knockout studies in the adult stage (unpublished data) show severe hydrocephalus phenotypes synonymous in MDS or lissencephaly patients, as seen in MRI. ...


... It is estimated to affect one in 100,000 live births [2]. Miller and Dieker contributed to the identification of MDS in 1963 and 1969, respectively, resulting in the nomenclature of the disease [15][16][17]. The genetic basis of the disorder has primarily been attributed to microdeletions along chromosome 17p13.3 ...


Miller–Dieker syndrome (MDS) is a rare genetic disorder characterized by congenital lissencephaly (absent or diminished cerebral gyri), facial dysmorphisms, neurodevelopmental retardation, intrauterine fetal demise, and death in early infancy or childhood. We present a case of a 4-year-old girl with MDS (17p13.3p13.2 deletion) who was admitted to the hospital due to fever and increased secretions from her nose, mouth, and tracheostomy tube (as she had been on a ventilator and G-tube dependent since birth). During the course of hospitalization, she developed multiorgan failure, third spacing, and significant lactic acidosis. The patient had a cardiorespiratory arrest and expired after 4 months and 8 days of hospitalization. We provide a synopsis of the main autopsy findings, with a focus on the neuropathologic anomalies.
... The median survival is less than 1 year. All survivors have profound mental retardation [108]. ...

This paper describes the contemporary state of knowledge regarding processes that regulate normal development of the embryonic–fetal central nervous system (CNS). The processes are described according to the developmental timetable: dorsal induction, ventral induction, neurogenesis, neuronal migration, post-migration neuronal development, and cortical organization. We review the current literature on CNS malformations associated with these regulating processes. We specifically address neural tube defects, holoprosencephaly, malformations of cortical development (including microcephaly, megalencephaly, lissencephaly, cobblestone malformations, gray matter heterotopia, and polymicrogyria), disorders of the corpus callosum, and posterior fossa malformations. Fetal ventriculomegaly, which frequently accompanies these disorders, is also reviewed. Each malformation is described with reference to the etiology, genetic causes, prenatal sonographic imaging, associated anomalies, differential diagnosis, complimentary diagnostic studies, clinical interventions, neurodevelopmental outcome, and life quality.
... The histopathology is best interpreted as forme fruste cobblestone malformation. Fusion of adjacent gyri and a minor degree of neuronal migration abnormalities were present; however, the extensive glioneuronal heterotopia of full-fledged cobblestone malformation (historically termed cobblestone lissencephaly) were not present (23,(33)(34)(35). Our patient manifested a mild degree of cerebellar neuronal migration abnormalities: focal failure of granule cell migration and subcortical heterotopia (Fig. 4). ...
Congenital muscular dystrophy type 1A (MDC1A) is caused by recessive variants in laminin α2 (LAMA2). Patients have been found to have white matter signal abnormalities on magnetic resonance imaging (MRI) but rarely structural brain abnormalities. We describe the autopsy neuropathology in a 17-year-old with white matter signal abnormalities on brain MRI. Dystrophic pathology was observed in skeletal muscle, and the sural nerve manifested a mild degree of segmental demyelination and remyelination. A diffuse, bilateral cobblestone appearance, and numerous points of fusion between adjacent gyri were apparent on gross examination of the cerebrum. Brain histopathology included focal disruptions of the glia limitans associated with abnormal cerebral cortical lamination or arrested cerebellar granule cell migration. Subcortical nodular heterotopia was present within the cerebellar hemispheres. Sampling of the centrum semiovale revealed no light microscopic evidence of leukoencephalopathy. Three additional MDC1A patients were diagnosed with cobblestone malformation on brain MRI. Unlike the autopsied patient whose brain had a symmetric distribution of cobblestone pathology, the latter patients had asymmetric involvement, most severe in the occipital lobes. These cases demonstrate that cobblestone malformation may be an important manifestation of the brain pathology in MDC1A and can be present even when patients have a structurally normal brain MRI.
... Earl Walker first described cases of lissencephaly in 1942 in reference to the "smooth brain" observed. Miller and Dieker contributed to the identification of MDS in 1963 and 1969 respectively, resulting in the nomenclature of the disease (Walker, 1942;Miller, 1963;Pilz and Quarrell, 1996). MDS results from contiguous gene deletion within 17p13.3 and mainly features cerebral agyria (absence of gyri), cerebral pachygyria (broad gyri), craniofacial deformities, and seizures (Barros Fontes et al., 2017). ...




Chromosome 17p13.3 is a region of genomic instability that is linked to different rare neurodevelopmental genetic diseases, depending on whether a deletion or duplication of the region has occurred. Chromosome microdeletions within 17p13.3 can result in either isolated lissencephaly sequence (ILS) or Miller-Dieker syndrome (MDS). Both conditions are associated with a smooth cerebral cortex, or lissencephaly, which leads to developmental delay, intellectual disability, and seizures. However, patients with MDS have larger deletions than patients with ILS, resulting in additional symptoms such as poor muscle tone, congenital anomalies, abnormal spasticity, and craniofacial dysmorphisms. In contrast to microdeletions in 17p13.3, recent studies have attracted considerable attention to a condition known as a 17p13.3 microduplication syndrome. Depending on the genes involved in their microduplication, patients with 17p13.3 microduplication syndrome may be categorized into either class I or class II. Individuals in class I have microduplications of the YWHAE gene encoding 14-3-3ε, as well as other genes in the region. However, the PAFAH1B1 gene encoding LIS1 is never duplicated in these patients. Class I microduplications generally result in learning disabilities, autism, and developmental delays, among other disorders. Individuals in class II always have microduplications of the PAFAH1B1 gene, which may include YWHAE and other genetic microduplications. Class II microduplications generally result in smaller body size, developmental delays, microcephaly, and other brain malformations. Here, we review the phenotypes associated with copy number variations (CNVs) of chromosome 17p13.3 and detail their developmental connection to particular microdeletions or microduplications. We also focus on existing single and double knockout mouse models that have been used to study human phenotypes, since the highly limited number of patients makes a study of these conditions difficult in humans. These models are also crucial for the study of brain development at a mechanistic level since this cannot be accomplished in humans. Finally, we emphasize the usefulness of the CRISPR/Cas9 system and next generation sequencing in the study of neurodevelopmental diseases.
... Classical lissencephaly can usually only be recognised on imaging after 28 weeks of pregnancy, as cerebral gyri are not fully developed prior to this gestation. [7][8][9] Miller-Dieker syndrome can be diagnosed antenatally by amniocentesis or chorionic villus sampling, when parents are found to have a balanced translocation. The risk of recurrence is extremely low in cases of de novo deletion. ...


... The importance for appropriate execution of the migration program in cortical projection neurons during brain development is highlighted in patients that suffer from neuronal migration disorders, such as isolated lissencephaly sequence (ILS) or Miller-Diecker Syndrome (MDS) (Dobyns and Truwit 1995 ;Gleeson and Walsh 2000 ;Guerrini and Parrini 2010 ;Pilz and Quarrell 1996 ;Ross and Walsh 2001 ). Lissencephaly is characterized by smooth brain surface, abnormal brain morphology and function; and lissencephaly patients suffer from mental retardation and epilepsy. ...

Coordinated migration of newly-born neurons to their target territories is essential for correct neuronal circuit assembly in the developing brain. Although a cohort of signaling pathways has been implicated in the regulation of cortical projection neuron migration, the precise molecular mechanisms and how a balanced interplay of cell-autonomous and non-autonomous functions of candidate signaling molecules controls the discrete steps in the migration process, are just being revealed. In this chapter, I will focally review recent advances that improved our understanding of the cell-autonomous and possible cell-nonautonomous functions of the evolutionarily conserved LIS1/NDEL1-complex in regulating the sequential steps of cortical projection neuron migration. I will then elaborate on the emerging concept that the Reelin signaling pathway, acts exactly at precise stages in the course of cortical projection neuron migration. Lastly, I will discuss how finely tuned transcriptional programs and downstream effectors govern particular aspects in driving radial migration at discrete stages and how they regulate the precise positioning of cortical projection neurons in the developing cerebral cortex.
... This term was proposed in humans to distinguish the flat brains of lower mammalian species and at present, several cases have been reported and different studies made in order to clarify its origin [1][2][3]5,6]. Two main forms of lissencephaly have been described: i) classical or type I lissencephaly, characterized by agyria (total loss of gyri) or pachygyria (few and broadened gyri) together with a very thick cortical gray matter layer, normal brainstem and grossly normal cerebellum or mild vermis hypoplasia, and ii) cobblestone or type II lissencephaly, in which affected children have an agyric brain, with a verrucous appearance and a less marked thickening of the cortex than in type I lissencephaly [1,6,7]. Microscopically, in type I lissencephaly the agyric cortex is mostly composed of four coarse layers: a molecular layer, an outer cellular layer, a sparsely cellular layer and a thick inner cellular layer [7,8]. ...
... Two main forms of lissencephaly have been described: i) classical or type I lissencephaly, characterized by agyria (total loss of gyri) or pachygyria (few and broadened gyri) together with a very thick cortical gray matter layer, normal brainstem and grossly normal cerebellum or mild vermis hypoplasia, and ii) cobblestone or type II lissencephaly, in which affected children have an agyric brain, with a verrucous appearance and a less marked thickening of the cortex than in type I lissencephaly [1,6,7]. Microscopically, in type I lissencephaly the agyric cortex is mostly composed of four coarse layers: a molecular layer, an outer cellular layer, a sparsely cellular layer and a thick inner cellular layer [7,8]. Type II lissencephaly is characterized by a complete disorganization of the cortex, without discernible layering, and by the presence of gliomesenchymal cell proliferations and neuroglial heterotopia in the leptomeninges [8]. ...
... In humans, although it is considered a rare disorder, lissencephaly is a well-recognized entity that belongs to the group of malformations caused by abnormal neuronal migration, with two main pathological syndromes described: type I (classical) and type II (cobblestone) [1,7,8]. According to the gross (absence of a verrucous appearance of the cortical surface) and microscopic findings, with no leptomeningeal neuronal heterotopia, the lissencephaly cases recorded in this study resemble the so-called type I or classical lissencephaly of humans. ...



Lissencephaly is a rare developmental brain disorder in veterinary and human medicine associated with defects in neuronal migration leading to a characteristic marked reduction or absence of the convolutional pattern of the cerebral hemispheres. In many human cases the disease has a genetic basis. In sheep, brain malformations, mainly cerebellar hypoplasia and forms of hydrocephalus, are frequently due to in utero viral infections. Although breed-related malformations of the brain have been described in sheep, breed-related lissencephaly has not been previously recorded in a peer reviewed publication.
Here we report neuropathological findings in 42 newborn lambs from a pure Churra breed flock, with clinical signs of weakness, inability to walk, difficulty in sucking and muscular rigidity observed immediately after birth. All the lambs showed near-total agyria with only a rudimentary formation of few sulci and gyri, and a severe cerebellar hypoplasia. On coronal section, the cerebral grey matter was markedly thicker than that of age-matched unaffected lambs and the ventricular system was moderately dilated. Histologically, the normal layers of the cerebral cortex were disorganized and, using an immunohistochemical technique against neurofilaments, three layers were identified instead of the six present in normal brains. The hippocampus was also markedly disorganised and the number and size of lobules were reduced in the cerebellum. Heterotopic neurons were present in different areas of the white matter. The remainder of the brain structures appeared normal. The pathological features reported are consistent with the type LCH-b (lissencephaly with cerebellar hypoplasia group b) defined in human medicine. No involvement of pestivirus or bluetongue virus was detected by immunohistochemistry. An analysis of pedigree data was consistent with a monogenic autosomal recessive pattern inheritance.
The study describes the clinical and pathological findings of lissencephaly with cerebellar hypoplasia in Churra lambs for which an autosomal recessive inheritance was the most likely cause. Histopathological features observed in the cerebral cortex and hippocampus are consistent with a possible failure in neuronal migration during brain development. This report suggests that lissencephaly should be considered in the differential diagnosis of congenital neurological disease in newborn lambs showing weakness, inability to walk and difficulty sucking.
... Patients have resultant craniofacial anomalies such as microcephaly with bitemporal hollowing, prominent forehead with vertical furrows, short nose with anteverted nares, thin vermilion border, and micrognathia [3]. There may be associated cardiac and renal defects, sacral dimples, joint contractures, and abnormal genitalia in males [7] . Developmental delay, seizures, failure to thrive, hypotonia, and feeding difficulties are common. ...
