Figure 1 - uploaded by Isabel Varela-Nieto
Content may be subject to copyright.

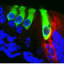
Anatomical description of the adult mouse inner ear: A. Lateral view of paint filled inner ear. Co, cochlea; V, vestibule; Asc, Lsc and Psc, anterior, lateral and posterior semicircular canals; Do, dorsal; Cd, caudal. B. Whole mounted cochlea showing the pigmented stria vascularis in the lateral wall and the round (RW) and oval (OW) windows. C. Midmodiolar section (inset) of the cochlea and the surrounding osseous otic capsule. Cochlear duct is divided in three fluid-filled scales: the scala vestibuli (SV), the scala media (SM) with the auditory receptor (black box) and the scala tympani (ST). D. Detail of a cochlear turn highlighting the spiral ganglion (SG, left black box) and the auditory receptor (organ of Corti, right black box). E. The organ of Corti contains the neurosensorial cells (IHC, OHC: inner and outer hair cells), the nonsensorial cells, the pillar cells (PC), the tectorial membrane (TM), the spiral limbus (SL) and the basilar membrane (BM). F. Phalloidin histochemistry of the organ of Corti, labeling F-actin in the stereocilia and cuticular plate of hair cells, the reticular lamina and pillar cells). G. Semi-thin section showing the cytoarchitecture of the spiral ganglion (SG). H. Electron micrograph of a spiral ganglionar cell (SGC), surrounded by Schwann cells (SC). I. Detail of the external compact (CM) and internal loose (LM) myelin sheaths in a SG Type I neuron. J. Detail of the marginal (MC), intermediate (IC) and basal (BC) cells in the stria vascularis. The spiral ligament (SpL) is close to the otic capsule. K. Midmodiolar section of the cochlea showing the Kir4.1 (KCNJ10, a K+ channel related with the production of the endocochlear potential) expression in the spiral ganglia (SG) and stria vascularis (StV). L-M. Detail of Kir4.1 (L) and Na+-K+-ATPase (M) expression in the stria vascularis (StV). Scale bars: A-C, K 0.5 mm; D 100 μm; E,F,J,L,M 50 μm; G 30 μm; H 5 μm; I 0.1 μm
Source publication








Sensorineural hearing loss is a clinical heterogeneous disorder and a significant health-care problem with tremendous socio-economic impact. According to WHO, "Over 5% of the world's population has disabling hearing loss -328 million adults and 32 million children-". In children, early hearing loss affects language acquisition. Hearing deficits are...
Contexts in source publication
Context 1
... gross morphology, cellular alterations, molecular mechanisms and hearing impairment has been studied in detail in a mutant mouse with a targeted knock-out mutation at the exon 4 of the Igf1 gene (Igf1 tm1Arge / Igf1 tm1Arge ). (88) The homozygous mutant mouse, when maintained in a hybrid background (129S/SvEv*MF1), presents delayed inner ear postnatal development, smaller cochlea and cochlear ganglia, significant decrease in the number and size of auditory neurons, abnormal otic capsule morphology, immature tectorial membrane, aberrant innervation patterns, increased neural apoptosis and deficits in myelination. (Table 2) (33,62,80) As a consequence, Igf1 -/-null mice develop a profound syndromic all- frequency bilateral sensorineural hearing loss with increased hearing threshold and delayed latencies of the auditory brainstem responses. (Figure 3) (35, 63, 101) The impact of IGF-I deficiency on the neuronal populations of the central auditory nuclei has not been yet ...
Context 2
... occurs when sound waves reach the structures inside the inner ear, where the sound wave vibrations are converted into nerve signals that the brain recognizes as sound. The ear consists of three major areas: outer ear, middle ear and inner ear. The inner ear is formed by fluid- filled canals and cavities, named the membranous labyrinth, which is encased into the bony labyrinth. It includes the auditory (hearing) and vestibular (balance) organs. The cochlea contains the sensory organ of Corti, where the highly specialized hair cells transform the mechanical input of the sound into an electrochemical signal that is conveyed to the brain.(1) The vestibule homes the balance receptors with specialized vestibular hair cells. (2) It is formed by five sensory organs, three cristae located at the base of the semicircular canals able to detect angular acceleration and two maculae that detect linear acceleration and gravity. (3) The sense of hearing and the maintenance of balance depend upon the functioning of sensory epithelia in the inner ear. Functional disturbance of this tissue leads respectively to hearing loss and/or disequilibrium resulting in dizziness and vertigo. Hearing and balance deficits are generally associated with the loss of the sensory "hair" cells and/or neurons by primary genetic defects or secondary to environmental factors including ototoxic damage, noise trauma, ageing or interactions between these factors ("A promenade round the cochlea" at http://www.neuroreille. com/promenade/english/corti/fcorti.htm). Figure 1 shows the anatomy of the cochlea that is formed by three fluid-filled coiled tubes around the modiolus. The scalas vestibuli and tympani are filled with perilymph, whereas the scala media is filled with endolymph and contains the hearing receptor. The organ of Corti is ...
Similar publications






Significance
Hair bundles are the sensory antennae that detect different types of mechanical signals in diverse sensory systems of vertebrates. Here we design and use a mechanical-load clamp to show that the mechanical properties of hair bundles and their accessory structures dictate their sensory behaviors. By demonstrating how the same organelle...
Citations
... 9,13 Also, IGF-1 receptors are expressed in the developing inner ear and the postnatal cochlear and vestibular ganglia. 11 In the inner ear, IGF-1 regulated the cell cycle and metabolism actions via intracellular signaling networks, RAF, AKT, and p38 MAPK protein kinases. 9 Mice lacking IGF-1 had increased auditory thresholds at early postnatal ages in mice and humans. ...
... 9 Mice lacking IGF-1 had increased auditory thresholds at early postnatal ages in mice and humans. 11 In the Igf1 null mouse, hearing loss is due to neuronal loss, poor innervation of the sensory hair cells, and age-related stria vascularis alterations. 9 In addition, IGF-1 serum levels decreased with aging and there are concomitant hearing loss and retinal degeneration in the mice. ...
The relationship between plasma glial cell-derived neurotrophic factor (GDNF) or insulin-like growth factor-1 (IGF-1) levels and age-related hearing impairment (ARHI) has not been reported in humans. By cross-sectional design, 268 subjects older than 33, with normal cognitive function and normal or symmetric sensorineural hearing loss, were selected randomly. Multivariate linear regression analysis was performed to test the impact of the plasma GDNF or IGF-1 level on the pure tone threshold of low frequencies (PTA-low) and high frequencies (PTA-high), respectively. Results showed that plasma GDNF and IGF-1 levels decreased with age without statistical significance. Multivariate linear regression analysis showed that GDNF or IGF-1 levels were not significantly correlated with PTA-low or PTA-high after adjusting age, gender, body mass index, systemic diseases, habits, and noise exposure. In conclusion, plasma GDNF or IGF-1 levels were not associated with the severity of ARHI in humans. However, these findings did not support the roles of GDNF or IGF-1 genotypes on hearing.
... But there are experimental data from animal studies (Rice & Barone 2000) consistent with human observational data suggesting an impact of early life factors on adult hearing function. Various mechanisms have been suggested for a direct causal impact of early life factors on hearing, including undernutrition affecting development (Barker 2004), alterations in gene expression (Egger et al. 2004;Provenzano & Domann 2007), the HPA-axis (Canlon et al. 2003), or growth factors Varela-Nieto et al. 2013). Early life factors have also been shown to impact susceptibility to Dawes et al. (Barker 2004). ...




Objectives: adverse prenatal and early childhood development may increase susceptibility of hearing loss in adulthood. The objective was to assess whether indices of early development are associated with adult-onset hearing loss in adults ≥18 years.
Design: in a systematic review and meta-analysis, four electronic databases were searched for studies reporting associations between indices of early development (birth weight and adult height) and adult-onset hearing loss in adults ≥18 years. We screened studies, extracted data, and assessed risk of bias. Authors were contacted to provide adjusted odds ratios from a logistic regression model for relationships between birth weight/adult height and normal/impaired hearing enabling a two-step individual patient data random-effects meta-analysis to be carried out. The study is registered with PROSPERO, CRD42020152214.
Results: four studies of birth weight and seven of adult height were identified. Three studies reported smaller birth weight associated with poorer adult hearing. Six studies reported shorter height associated with poorer hearing. Risk of bias was low to moderate. Four studies provided data for two-step individual patient data random-effects meta-analysis. Odds of hearing impairment were 13.5% lower for every 1 kg increase in birth weight [OR: 0.865 (95% confidence interval: 0.824 to 0.909)] in adulthood over two studies (N=81,289). Every 1 cm increase in height was associated with a 3% reduction in the odds of hearing impairment [OR: 0.970 (95% confidence interval: 0.968 to 0.971)] over four studies (N=156,740).
Conclusions: emerging evidence suggests that adverse early development increases the likelihood of hearing impairment in adulthood. Research and public health attention should focus on the potential for prevention of hearing impairment by optimizing development in early life.
... But there are experimental data from animal studies (Rice & Barone 2000) consistent with human observational data suggesting an impact of early life factors on adult hearing function. Various mechanisms have been suggested for a direct causal impact of early life factors on hearing, including undernutrition affecting development (Barker 2004), alterations in gene expression (Egger et al. 2004;Provenzano & Domann 2007), the HPA-axis (Canlon et al. 2003), or growth factors Varela-Nieto et al. 2013). Early life factors have also been shown to impact susceptibility to Dawes et al. (Barker 2004). ...

Objectives:
Adverse prenatal and early childhood development may increase susceptibility of hearing loss in adulthood. The objective was to assess whether indices of early development are associated with adult-onset hearing loss in adults ≥18 years.
Design:
In a systematic review and meta-analysis, four electronic databases were searched for studies reporting associations between indices of early development (birth weight and adult height) and adult-onset hearing loss in adults ≥18 years. We screened studies, extracted data, and assessed risk of bias. Authors were contacted to provide adjusted odds ratios from a logistic regression model for relationships between birth weight/adult height and normal/impaired hearing enabling a two-step individual patient data random-effects meta-analysis to be carried out. The study is registered with PROSPERO, CRD42020152214.
Results:
Four studies of birth weight and seven of adult height were identified. Three studies reported smaller birth weight associated with poorer adult hearing. Six studies reported shorter height associated with poorer hearing. Risk of bias was low to moderate. Four studies provided data for two-step individual patient data random-effects meta-analysis. Odds of hearing impairment were 13.5% lower for every 1 kg increase in birth weight [OR: 0.865 (95% confidence interval: 0.824 to 0.909)] in adulthood over two studies (N=81,289). Every 1 cm increase in height was associated with a 3% reduction in the odds of hearing impairment [OR: 0.970 (95% confidence interval: 0.968 to 0.971)] over four studies (N=156,740).
Conclusions:
Emerging evidence suggests that adverse early development increases the likelihood of hearing impairment in adulthood. Research and public health attention should focus on the potential for prevention of hearing impairment by optimizing development in early life.
... In animal and human models, inborn IGF-1 deficiency results in a phenotype characterized by profound hearing loss, cognitive deficits, and short stature. 44 Based on this and its role as a neuroprotective agent, a decline in IGF-1 levels has been implicated in the progression of ARHL and, either as a result of hearing loss or through a direct influence on the nervous system, a decline in cognitive performance. 45 Unlike other sensory deficits, hearing loss may be the most modifiable. ...

This review examines the relationship between cochlear implantation and cognition and quality of life in older adults, as well as how frailty affects outcomes for older patients with cochlear implants. A growing body of evidence suggests that there is a strong association between hearing loss and cognitive impairment. Preliminary studies suggest that cochlear implantation in older adults may be protective against cognitive decline. While studies have observed a positive impact of cochlear implantation on quality of life, currently it is unclear what factors contribute the most to improved quality of life. Frailty, as a measurement of general health, likely plays a role in complication rates and quality-of-life outcomes after cochlear implantation, though larger prospective studies are required to further elucidate this relationship.
... Human insulin-like growth factor 1 (IGF-1) homozygous deficiency is an ultrarare disease associated with dwarfism, mental retardation, and syndromic sensorineural hearing loss (SNHL) [4]. However, IGF-1 heterozygous mutations have not been associated with congenital SNHL yet [5][6][7]. ...


Insulin-like growth factor 1 (IGF-1) deficiency is an ultrarare syndromic human sensorineural deafness. Accordingly, IGF-1 is essential for the postnatal maturation of the cochlea and the correct wiring of hearing in mice. Less severe decreases in human IGF-1 levels have been associated with other hearing loss rare genetic syndromes, as well as with age-related hearing loss (ARHL). However, the underlying mechanisms linking IGF-1 haploinsufficiency with auditory pathology and ARHL have not been studied. Igf1-heterozygous mice express less Igf1 transcription and have 40% lower IGF-1 serum levels than wild-type mice. Along with ageing, IGF-1 levels decreased concomitantly with the increased expression of inflammatory cytokines, Tgfb1 and Il1b, but there was no associated hearing loss. However, noise exposure of these mice caused increased injury to sensory hair cells and irreversible hearing loss. Concomitantly, there was a significant alteration in the expression ratio of pro- and anti-inflammatory cytokines in Igf1+/− mice. Unbalanced inflammation led to the activation of the stress kinase JNK and the failure to activate AKT. Our data show that IGF-1 haploinsufficiency causes a chronic subclinical proinflammatory age-associated state and, consequently, greater susceptibility to stressors. This work provides the molecular bases to further understand hearing disorders linked to IGF-1 deficiency.
... Noteworthy, all of these players are also involved, at some level, in ear development and human deafness [10][11][12][13][14][15]. Finally, the RFX transcription factors, which are crucial for hearing in mice, and the Rbm24 RNA-binding protein, which is expressed in the mechanosensory cells of the auditory/vestibular system during vertebrate development, positively regulate Smpx expression [16,17]. ...


The last decade has witnessed the identification of several families affected by hereditary non-syndromic hearing loss (NSHL) caused by mutations in the SMPX gene and the loss of function has been suggested as the underlying mechanism. In the attempt to confirm this hypothesis we generated an Smpx-deficient zebrafish model, pointing out its crucial role in proper inner ear development. Indeed, a marked decrease in the number of kinocilia together with structural alterations of the stereocilia and the kinocilium itself in the hair cells of the inner ear were observed. We also report the impairment of the mechanotransduction by the hair cells, making SMPX a potential key player in the construction of the machinery necessary for sound detection. This wealth of evidence provides the first possible explanation for hearing loss in SMPX-mutated patients. Additionally, we observed a clear muscular phenotype consisting of the defective organization and functioning of muscle fibers, strongly suggesting a potential role for the protein in the development of muscle fibers. This piece of evidence highlights the need for more in-depth analyses in search for possible correlations between SMPX mutations and muscular disorders in humans, thus potentially turning this non-syndromic hearing loss-associated gene into the genetic cause of dysfunctions characterized by more than one symptom, making SMPX a novel syndromic gene.
... OPEN 1 (PKB) pathways, which potentially leads to cell proliferation or cell differentiation 10 . IGF-1 functions are critical for the protection of nerve cells against neurodegenerative processes 11,12 . Deficiency in the IGF-1 gene in humans was associated with neuronal disorders such as microcephaly, mental retardation, and bilateral sensorineural deafness [13][14][15] . ...



Insulin-like Growth Factor-1 (IGF-1) is involved in the normal development and survival of retinal cells. Low-density lipoprotein Receptor-related Protein-1 (LRP1) plays a key role on the regulation of several membrane proteins, such as the IGF-1 receptor (IGF-1R). In brain astrocytes, LRP1 interact with IGF-1R and the glucose transporter type 1 (GLUT1), regulating the glucose uptake in these cells. Although GLUT1 is expressed in retinal Müller Glial Cells (MGCs), its regulation is not clear yet. Here, we investigated whether IGF-1 modulates GLUT1 traffic to plasma membrane (PM) and glucose uptake, as well as the involvement of LRP1 in this process in the human Müller glial-derived cell line (MIO-M1). We found that IGF-1 produced GLUT1 translocation to the PM, in a time-dependent manner involving the intracellular signaling activation of MAPK/ERK and PI3K/Akt pathways, and generated a significant glucose uptake. Moreover, we found a molecular association between LRP1 and GLUT1, which was significantly reduced by IGF-1. Finally, cells treated with specific siRNA for LRP1 showed an impaired GLUT1 expression on PM and decreased glucose uptake induced by IGF-1. We conclude that IGF-1 regulates glucose homeostasis in MGCs involving the expression of LRP1.
... Syndromic HL is a common condition in many RDs including insulin-like growth factor I (IGF-1) deficiency, an ultra-RD caused by homozygous mutations in IGF1 and associated with growth retardation, intellectual deficit, and HL (Varela-Nieto et al., 2013). The use of experimental models is practically the only way to investigate the pathology of ultra-RDs. ...

Animal models are invaluable for biomedical research, especially in the context of rare diseases, which have a very low prevalence and are often complex. Concretely mouse models provide key information on rare disease mechanisms and therapeutic strategies that cannot be obtained by using only alternative methods, and greatly contribute to accelerate the development of new therapeutic options for rare diseases. Despite this, the use of experimental animals remains controversial. The combination of respectful management, ethical laws and transparency regarding animal experimentation contributes to improve society’s opinion about biomedical research and positively impacts on research quality, which eventually also benefits patients. Here we present examples of current advances in preclinical research in rare diseases using mouse models, together with our perspective on future directions and challenges.
... The main clinical features of a severe homozygous IGF1 defect include extreme pre-and post-natal growth failure, poor feeding, severe microcephaly, retrognathia, sensorineural deafness and severe global developmental delay (Table 1) . The finding of sensorineural deafness is consistent with the observations in postnatal IGF-I deficient mice, which show delayed inner ear maturation and neuronal loss (Camarero et al., 2001;Varela-Nieto et al., 2013). Birth length SDS varies between − 4.3 and − 6.5, much shorter than in patients with complete GH deficiency or insensitivity. ...

The insulin-like growth factor (IGF) system comprises two ligands, IGF-I and IGF-II, that regulate multiple physiological processes, including mammalian development, metabolism and growth, through the type 1 IGF receptor (IGF-1R). The growth hormone (GH)-IGF-I axis is the major regulator of longitudinal growth. IGF-II is expressed in many tissues, notably the placenta, to regulate human pre- and post-natal growth and development. This review provides a brief introduction to the IGF system and summarizes findings from reports arising from recent larger genomic sequencing studies of human genetic mutations in IGF1 and IGF2 and genes of proteins regulating IGF action, namely the IGF-1R, IGF-1R signaling pathway components and the IGF binding proteins (IGFBPs). A perspective on the effect of homozygous mutations on structure and function of the IGFs and IGF-1R is also given and this is related to the effects on growth.
... When necessary, corrective eyes saccades (quick, simultaneous movement of both eyes in the same direction) keep the image of the object of interest in the fovea, another important step to associate movement with balance [5,6]. Hearing and balance deficits are often caused by the loss of the sensory hair cells or neurons by genetic, acquired cause, or by aging [7]. ...
... Not surprisingly, congenital disruption of IGF-I signaling in mice results into an abnormal development of the cochlea [8]. Data of the vestibular function of IGF-I in postnatal life in mice are scanty [7] and in men are lacking. ...
... GH receptor knockout mice seem not to exhibit gross problems with their sense of balance in various tests of physical performance [14,26]. Nevertheless, IGF-I-null mice exhibit a moderate vestibular phenotype, with alterations in the contact righting test, trunk curl test, and swimming test [7]. Although a different orientation of the cochlea in relation to the brain may occur, a change in the vestibular sensory epithelium is also possible in these rodents [27]. ...




Purpose:
Body balance involves the vestibular, visual, and proprioceptive systems. IGF-I is a GH-dependent key factor in the development and postnatal differentiation of the inner ear in mice and men, but its role in the vestibular function in adult humans is unknown. We have previously described a cohort of individuals with severe isolated GH deficiency (IGHD) caused by a mutation in the GHRH receptor (GHRHR) gene. These individuals complain of dizziness, exhibit mild sensorineural loss, but have normal postural balance, without increase in falls risk. The aim of this study was to evaluate their vestibular function.
Methods:
We performed physical examination (clinical head impulse and Fukuda dynamic stepping test), oculomotor (saccadic eye movements, spontaneous, semi-spontaneous and opotokinetic nystagmus, and pendular tracking) and caloric stimulation (postcaloric reflex and ocular fixation index) tests, in 15 GH-naïve IGHD (seven males) and 15 controls (five males).
Results:
IGHD subjects showed lower height and weight, with similar BMI to controls, and higher number of individuals with abnormal clinical head impulse test and abnormal oculomotor tests, namely the saccadic movements and the spontaneous nystagmus. There was a nonsignificant trend in abnormalities in the Fukuda stepping test and postcaloric reflex test.
Conclusions:
Adult untreated IGHD individuals have higher prevalence of moderate peripheral vestibular impairment, and of abnormal vestibular-ocular reflex.